On supposera tout au long de cette fiche que le système étudié est fermé, de volume constant et de composition uniforme.
b. Rappel sur l'avancement d'une réaction
Soit la réaction générale qu'on peut noter
L'avancement de la réaction à la date t est
Avec = quantité de matière de à l'instant et quantité de matière de à l'instant .
c. Vitesse de réaction
La vitesse de réaction s'exprime de la façon suivante :
Finalement,
On peut ausi définir la vitesse d'apparition de :
et la vitesse de disparition de s'écrit
2. Facteurs cinétiques
Les principaux facteurs influençant la vitesse de réaction sont les suivants :
La température : la plupart du temps, la vitesse de réaction augmente si la température augmente (il y a quelques exceptions);
Les concentrations ;
Etat physique des corps, solvant, radiations lumineuses ;
Catalyse : un catalyseur est une espèce chimique capable d'accélérer une réaction thermodynamiquement possible sans subir de changement permanent. Sauf auto-catalyse, il n'apparaît pas dans l'équation-bilan de la réaction.
3. Méthodes expérimentales d'étude
a. Méthodes chimiques
On fait le dosage de la totalité du mélange ou d'un prélèvement. Il faut bloquer la réaction dont on étudie la cinétique pendant le dosage :
soit on effectue une trempe (= refroidissement brutal),
soit on dilue fortement le système réactionnel,
soit on ajoute un corps faisant disparaître l'un des réactifs par une réaction rapide.
b. Méthodes physiques
Les principales méthodes physiques employées sont les suivantes :
Mesure de pression ;
Mesure spectroscopique (réviser les notions d'absorbance et la loi de Beer-Lambert) ;
Mesure électrique : pH, potentiel d'électrode, conductivité (réviser cette notion).
4. Notion expérimentale d'ordre
a. Définition
Soit la réaction
Dans d'assez nombreux cas, l'expérience indique que la vitesse de réaction peut être mise sous la forme :
les coefficients , ... étant entiers ou fractionnaires, étant une constante, appelée constante de vitesse.
= ordre partiel par rapport au réactif et = ordre global de la réaction.
b. Dégénérescence de l'ordre
Si on met le réactif en excès par rapport à tous les autres réactifs, il sera relativement peu consommé :
d'où avec = constante de vitesse apparente.
L'ordre global passe de à = ordre apparent.
Pour
avec , , entiers les plus petits possibles,
si , on dit que la réaction suit la loi de Van't Hoffc'est le cas des réactions élémentaires dont l'équation-bilan représente le mécanisme mais attention : la réciproque n'est pas vraie.
c. Influence de la température
Loi d'Arrhénius :
Avec : ; .
= énergie d'activation de la réaction (en J/mol) indépendante de T.
II. Cinétique formelle
1. Rappels
Lors d'une réaction chimique, des réactifs sont mis en jeu donnant un ou plusieurs produits.
Une réaction se fait donc à une certaine vitesse.
Il existe la vitesse de formation du produit et la vitesse de disparition du réactif.
L'équation-bilan simplifiée sera considérée par la suite : A + B C + D
2. Vitesses
- Vitesse de formation : Vf(t) = + en mol/s, mol/min, ...
- Vitesse de disparition : Vd(t) = - en mol/s, mol/min, ... Avec : n le nombre de moles du produit formé ou du réactif disparu
- Relation : x VdA = x VdV = x VfC = x VfD
- Vitesse moyenne de formation : =
- Vitesse moyenne de dispariton: =
- L'avancement : = = = = en moles
- Vitesse de réaction :
A +
B
C
D
à t=0
nA0
nB0
0
0
à t
nA0-
nB0-
Donc
* nA = na0 - = 0 - =
Remarque : On peut procéder pareil pour les nombres de moles nB
* nc = = =
Remarque : On peut procéder pareil pour les nombres de moles nD
- Vitesse volumique (ou spécifique): C'est la vitesse de réaction a volume constant.
On la note Vs.
Vs =
= en mol/L/s, mol/L/min, ...
D'après la formule, Vs, la vitesse volumique, est donc le rapport de la vitesse sur le volume.
Soit Vs = x
= x
= x
= x
3. Loi de vitesse
La loi de vitesse est une relation mathématique entre la vitesse de réaction et les concentration des reactifs A et B.
Vs = k x [A]a x [B]b
avec :
k, une constante de vitesse (dépendant de la température et de la nature de la réaction). [A], la concentration du réactif A en mol/L. [B], la concentration du réactif B en mol/L. a, l'ordre partiel de A, un nombre sans unité. b, l'ordre partiel de B, un nombre sans unité.
On note a + b = N, un nombre sans unité, l'ordre global de la réaction.
Remarque : k a une unité qui est à déterminer selon les cas
- Si N = 0, a=0 et b=0, k est donc en L-1.s-1 (On procède par homogénéité)
- Si N = 1, a=1 et b=0, k est donc en s-1
- Si N = 2, a=1 et b=1, k est donc en L.mol-1.s-1
Ce sont l'ordre 0, l'ordre premier et l'ordre second de la réaction.
La vitesse de cette loi cinétique peut être différente pour deux réactions, dont les réactifs sont identiques, selon divers facteurs cinétiques :
* La concentration des réactifs ou la pression pour des gaz
* La température du milieu
* L'éclairement
* Le contact entre les réactifs
* La nature du solvant
* Un catalyseur, initiateur, inhibiteur ou amorceur
4. Temps de demi-réaction et équation de droite
Le temps de demi réaction est le temps au bout duquel la moitier de la concentration initiale en réactif a été consommée. Elle se note t1/2.
Rappel : Une droite se note f(x) = a x + b où :
a est le coefficient directeur de la droite.
x est la variable (ici, ce sera t, le temps)
b est l'ordonnée a l'origine (la valeur de y lorsqu'on sera a t=0)
Le but étant d'observer la concentration d'un réactif en fonction du temps lors de la réaction, il faut isoler le plus possible la concentration
tout en ayant une équation de droite.
On considere ici une réaction de la forme
A B
* Si N = 0
a=0 Vs = k x [A]0 = k
Et [A] = [A]0 -
= 0 -
=
= Vs = k
d[A] = -k dt
[A]0[A] d[A] =
-k 0t dt
[A] - [A]0 = -kt
[A] = -k t + [A]0
Temps de demi réaction : [A] = = -kt1/2 + [A]0 t1/2 =
* Si N = 1
a=1 Vs = k x [A]1 = k [A]
Et [A] = [A]0 -
* Si N = 2
a=2 Vs = k x [A]² = k [A][A]
Et [A] = [A]0 -
= 0 -
=
= Vs = k [A]
= -k dt
[A]0[A] -
= k 0t dt
- = kt
= k t +
Temps de demi réaction : [A] = - = kt1/2 = kt1/2 t1/2 =
Avec plusieurs réactifs, on suit sur le modele de
A + B C + D
* Si B est en excès et [B]0 très grand
On considerer [B] = constante
On note donc k[B]b, une pseudo constante, par kapp
Ainsi : Vs = kapp[A]a
* Si A est en excès et [A]0 très grand
On considerer [A] = constante
On note donc k[A]a, une pseudo constante, par kapp
Ainsi : Vs = kapp[B]b
5. Les types réactionels
Une réaction complexes, a la difference des réactions élementaires, est composée de plusieurs transitions.
Il existe trois types de réactions complexes :
Les réactions successives du type A B C
En passant de A à B et de B à C par deux constantes k1 et k2 différentes
Les réactions successives pré-équilibrées du type A + B I C
Avec I, un état intermediaire non stable.
Ici, on passe de A+B à I par la constante k1, de I à A+B par k-1 et de I à C par k2
Les réactions parallèles :
*Compétitives : de la forme A + B C + D et A + B' C + D'
*Jumelles : de la forme A + B C + D et A + B C' + D'
Publié par gbm, relue par gts2/vanoise
le
ceci n'est qu'un extrait
Pour visualiser la totalité des cours vous devez vous inscrire / connecter (GRATUIT) Inscription Gratuitese connecter
Merci à Saphirite pour avoir contribué à l'élaboration de cette fiche
Désolé, votre version d'Internet Explorer est plus que périmée ! Merci de le mettre à jour ou de télécharger Firefox ou Google Chrome pour utiliser le site. Votre ordinateur vous remerciera !
 qu'on peut noter
qu'on peut noter 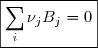
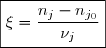
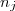 = quantité de matière de
= quantité de matière de 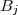 à l'instant
à l'instant 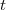 et
et 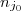 quantité de matière de
quantité de matière de 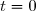 .
.
}{dt} = \dfrac{1}{\nu _j} . \dfrac{d(\frac{n_j}{V})}{dt})
![\boxed{v = \dfrac{1}{\nu _j} . \dfrac{d [B_j]}{dt}}](https://latex.ilephysique.net/latex-0.tex?\boxed{v = \dfrac{1}{\nu _j} . \dfrac{d [B_j]}{dt}})
![v_{app} = + \dfrac{d [B_j]}{dt}](https://latex.ilephysique.net/latex-0.tex?v_{app} = + \dfrac{d [B_j]}{dt})
![v_{disp} = - \dfrac{d [B_j]}{dt}](https://latex.ilephysique.net/latex-0.tex?v_{disp} = - \dfrac{d [B_j]}{dt})
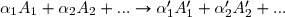
![\boxed{v = k.[A_1]^{p_1}.[A_2]^{p_2} ...[A_m]^{p_m}}](https://latex.ilephysique.net/latex-0.tex?\boxed{v = k.[A_1]^{p_1}.[A_2]^{p_2} ...[A_m]^{p_m}})
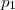 ,
, 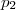 ...
... 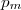 étant entiers ou fractionnaires,
étant entiers ou fractionnaires, 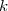 étant une constante, appelée constante de vitesse.
étant une constante, appelée constante de vitesse.
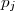 = ordre partiel par rapport au réactif
= ordre partiel par rapport au réactif 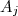 et
et 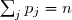 = ordre global de la réaction.
= ordre global de la réaction.
![[A_j] \approx constante](https://latex.ilephysique.net/latex-0.tex?[A_j] \approx constante)
![v = k.[A_1]^{p_1}... \textcolor{blue}{[A_j]^{p_j}} ...[A_m]^{p_m}= k' . [A_1]^{p_1} ...[A_m]^{p_m}](https://latex.ilephysique.net/latex-0.tex?v = k.[A_1]^{p_1}... \textcolor{blue}{[A_j]^{p_j}} ...[A_m]^{p_m}= k' . [A_1]^{p_1} ...[A_m]^{p_m}) avec
avec ![k' = k.[A_j]^{p_j}](https://latex.ilephysique.net/latex-0.tex?k' = k.[A_j]^{p_j}) = constante de vitesse apparente.
= constante de vitesse apparente.
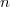 à
à ) = ordre apparent.
= ordre apparent.
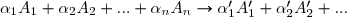 avec
avec 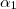 ,
, 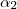 ,
, 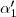 entiers les plus petits possibles,
si
entiers les plus petits possibles,
si ![v = k.[A_1]^{\alpha _1} ...[A_n]^{\alpha _n}](https://latex.ilephysique.net/latex-0.tex?v = k.[A_1]^{\alpha _1} ...[A_n]^{\alpha _n}) , on dit que la réaction suit la loi de Van't Hoff
, on dit que la réaction suit la loi de Van't Hoff 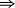 c'est le cas des réactions élémentaires dont l'équation-bilan représente le mécanisme mais attention : la réciproque n'est pas vraie.
c'est le cas des réactions élémentaires dont l'équation-bilan représente le mécanisme mais attention : la réciproque n'est pas vraie.
 = - \dfrac{E_a}{R.T} \Leftrightarrow \boxed{\dfrac{d \ln(k)}{dT} = \dfrac{E_a}{R.T^2}})
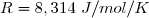 ;
; 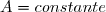 .
.
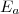 = énergie d'activation de la réaction (en J/mol) indépendante de T.
= énergie d'activation de la réaction (en J/mol) indépendante de T.
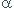 A +
A + 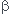 B
B 
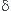 C +
C + 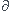 D
D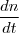 en mol/s, mol/min, ...
en mol/s, mol/min, ...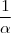 x VdA =
x VdA = 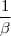 x VdV =
x VdV = 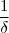 x VfC =
x VfC = 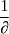 x VfD
x VfD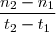 =
= 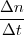
}{t_2-t_1}}) =
= 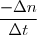
 =
= 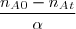 =
= 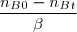 =
= }{\delta}}) =
= }{\partial}}) en moles
en moles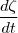

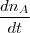 = 0 -
= 0 - 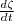
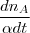
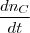 =
= 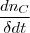
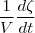 =
= 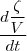 en mol/L/s, mol/L/min, ...
en mol/L/s, mol/L/min, ...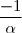 x
x ![{\dfrac{d[A]}{dt}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[A]}{dt}}) =
= 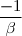 x
x ![{\dfrac{d[B]}{dt}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[B]}{dt}}) =
= 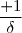 x
x ![{\dfrac{d[C]}{dt}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[C]}{dt}}) =
= 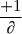 x
x ![{\dfrac{d[D]}{dt}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[D]}{dt}})

![{\dfrac{-d [A]}{\alpha dt}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{-d [A]}{\alpha dt}}) = Vs = k
= Vs = k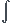 [A]0[A] d[A] =
-
[A]0[A] d[A] =
-![{\dfrac{[A]_0}{2}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{[A]_0}{2}})
![{\dfrac{[A]_0}{2\alpha k}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{[A]_0}{2\alpha k}})
![{\dfrac{d[A]}{[A]}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[A]}{[A]}}) = -
= -![{\frac{d[A]}{[A]}}](https://latex.ilephysique.net/latex-0.tex?{\frac{d[A]}{[A]}}) = -
= -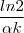
![{\dfrac{d[A]}{[A]²}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[A]}{[A]²}}) = -
= -![{\dfrac{d[A]}{[A]^2}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{d[A]}{[A]^2}}) =
= ![{\dfrac{1}{[A]}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{1}{[A]}}) -
- ![{\dfrac{1}{[A]_0}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{1}{[A]_0}}) =
= ![{\dfrac{1}{\frac{[A]_0}{2}}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{1}{\frac{[A]_0}{2}}}) -
- ![{\dfrac{1}{[A]_0 ak}}](https://latex.ilephysique.net/latex-0.tex?{\dfrac{1}{[A]_0 ak}})
 I
I  Chimie en post-bac
Chimie en post-bac