Ce document est la suite logique de celui concernant le premier principe.
Il s'agit toujours de l'étude de la thermodynamique classique d'un système fermé.
Sauf cas particulier clairement précisé ultérieurement, on considère que seules les forces de pression sont susceptibles de travailler.
Quelques exercices d'illustration avec leurs corrigés sont proposés.
1. Objectif
Le premier principe permet de déterminer des transferts d'énergie en utilisant les propriétés des fonctions d'état U et H.
On peut maintenant se poser la question : tous les transferts d'énergie théoriquement envisageables sont-ils possibles en pratique ?
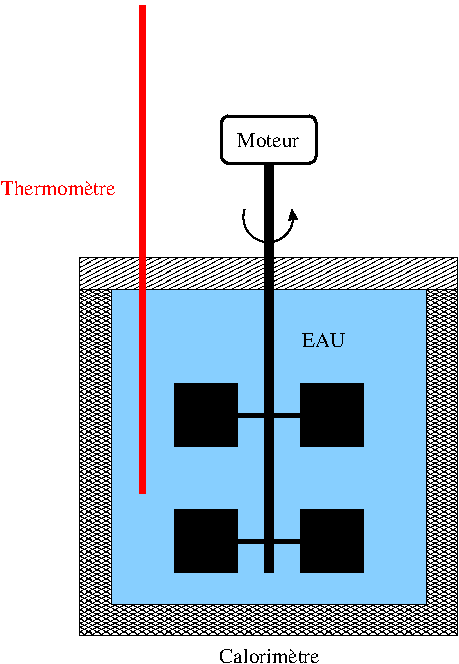
Prenons un exemple simple : une certaine quantité d'eau est enfermée dans un calorimètre supposé parfaitement calorifugé (parois athermanes).
Un agitateur est relié à un moteur.
La rotation des pales de l'agitateur provoque une augmentation de l'agitation thermique de l'eau, donc une augmentation de son énergie interne, ce qui se traduit par une augmentation de température de l'eau.
On peut dire que l'augmentation de l'énergie interne de l'eau est égale au travail reçu par l'eau, ce travail étant le travail des forces exercées par les pales en mouvement sur l'eau :
On pourrait envisager l'évolution inverse : le moteur étant enlevé, le refroidissement adiabatique de l'eau provoquerait la rotation des pales, l'eau perdant alors de l'énergie interne en fournissant du travail à l'extérieur.
Cela correspondrait à :
L'expérience montre que cette évolution inverse est impossible !
Manque donc à la thermodynamique un deuxième principe, capable de prédire si une évolution est possible ou non.
C'est ce « principe d'évolution » que nous allons étudier ici.
Nous montrerons, paragraphe 7.2 la cohérence de ce nouveau principe avec les faits expérimentaux.
Depuis que l'étude de la thermodynamique existe, motivée au départ par la mise au point et la compréhension des machines thermiques (machines à vapeur, moteurs thermiques, etc.) plusieurs énoncés de ce principe se sont succédés.
Nous retenons celui le plus fréquemment enseigné actuellement : celui de ILYA PRIGOGINE, chimiste belge d'origine russe, prix Nobel en 1977.
2. Notion de réversibilité
Définition
Une transformation est qualifiée de réversible lorsqu'elle possède les deux propriétés suivantes :
1° : elle est quasi statique ;
2° : elle est renversable.
Le qualificatif « quasi statique » a été largement développé au paragraphe 3.4 de la fiche sur le premier principe.
La transformation doit être suffisamment lente pour que le système puisse être à chaque instant considéré en état d'équilibre interne :
la température, à un instant donnée, est la même en tout point du système ; même chose pour la pression.
Le système vérifie ainsi une équation d'état à chaque instant.
De plus, si le système est en contact thermique avec le milieu extérieur (parois diathermanes), on doit avoir à chaque instant : }.
Si le système est séparé de l'extérieur par une parois mobile (piston dans un cylindre par exemple), on doit avoir à chaque instant : .
Une transformation d'un état 1 à un état 2 est « renversable » s'il est possible de revenir de l'état 2 à l'état 1 par la même succession d'états intermédiaires (par le même chemin décrit en sens inverse).
Certaines transformations peuvent être quasi statiques sans être réversibles. Prenons deux exemples.
Exemple 1
Il concerne la thermodynamique des solides et non des fluides.
Il fait intervenir des forces autres que celles de pression mais on, le cite néanmoins en raison de son caractère particulièrement démonstratif.
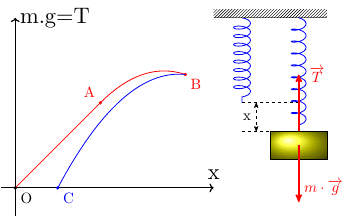
On considère un ressort vertical dont l'extrémité supérieure est fixée à un support immobile.
À l'extrémité inférieure on fixe de très petites masselottes ;
on attend entre chaque ajout que l'équilibre soit atteint, la tension T du ressort devenant égale à m.g et son augmentation de longueur étant « x ».
Cet allongement du ressort est ainsi quasi statique.
Peut-on le considérer comme renversable ? Deux situations sont envisageables :
Premier cas : l'état final ne dépasse pas la limite d'élasticité du ressort (état final sur la courbe rouge entre O et A) : pour chaque état intermédiaire, on peut écrire : où est la constante de raideur du ressort. Si on enlève progressivement et lentement les masselottes, le ressort va retrouver son état initial correspondant au point O, tous les états intermédiaires étant encore sur le segment de droite rouge. Bref : le ressort revient à son état initial par la même succession d'états intermédiaires. La transformation est donc renversable. Puisqu'elle est aussi quasi statique, elle est réversible.
Deuxième cas : lors de l'ajout très progressif des masselottes, l'état final dépasse la limite d'élasticité sans atteindre l'état de rupture du ressort et correspond au point B sur la courbe caractéristique. Si on enlève progressivement et lentement les masselottes, les états intermédiaires sont toujours des états d'équilibre mais ils correspondent à la courbe bleue sur le schéma. Le ressort ne retrouve pas son état initial pour m=0 ; l'état final correspond au point C sur la courbe : le ressort est plus long qu'à l'état initial. La transformation n'est donc pas renversable. Bien que quasi statique, elle n'est donc pas réversible.
Exemple 2
Il concerne l'ensemble des évolutions où interviennent des forces de frottements : forces de frottement exercées par la parois d'un cylindre sur un piston par exemple. Partant d'un état d'équilibre imaginons un déplacement élémentaire dx du piston qui provoque un travail élémentaire W, un transfert thermique Q, une variation élémentaire d'énergie interne du gaz dU, etc. Si la transformation est renversable, inverser le sens du mouvement, soit inverser le signe de dx, doit inverser les signes de W, de Q et de dU en conservant leurs valeurs absolues. Ce remplacement de W par (-W) est impossible si les forces de frottement interviennent dans l'expression du travail élémentaire ; en effet : le travail des forces de frottement est toujours négatif quel que soit le sens du mouvement. Ainsi, en présence de frottements, l'évolution peut être quasi statique si elle est très lente mais elle ne peut pas être renversable. La présence de frottements interdit la réversibilité.
3. Énoncé du deuxième principe de la thermodynamique
Deuxième principe de la thermodynamique
Ce principe postule l'existence d'une nouvelle fonction d'état extensive du système fermé, l'entropie, notée S.
La variation élémentaire d'entropie est une somme de deux termes :
1° : un terme d'échange, dont l'expression, en cas d'échange thermique avec une source de température Text, s'écrit :
(1)
2° : un terme de création, nul en cas d'évolution réversible et strictement positif dans les autres cas :
(2)
Ce qui revient à écrire la variation élémentaire d'entropie sous la forme :
(3)
Cette relation peut s'intégrer entre un état initial 1 et un état final 2 du système fermé :
(4)
Les bornes d'intégration dépendent du type d'évolution ;
l'expression n'est pas nécessairement simple si Text n'est pas fixe lors de l'évolution.
L'entropie de création est a priori inconnue ;
on sait seulement qu'elle ne peut pas être négative.
Cet énoncé appelle plusieurs remarques importantes :
1° : En cas d'évolution spontanée d'un système isolé thermiquement, la transformation est à la fois adiabatique et irréversible.
L'entropie d'échange est nécessairement nulle et l'entropie de création nécessairement positive : .
En pratique, il est toujours possible de choisir un système suffisamment étendu pour qu'il soit isolé thermiquement (dans les cas extrêmes, on peut choisir l'univers entier !).
Nous avons donc bien dans ce principe un principe d'évolution : une évolution spontanée d'un système thermiquement isolé se fait toujours dans le sens qui augmente l'entropie.
2° : Dans le cas limite d'une évolution réversible, l'entropie de création est nulle et l'évolution est quasi statique : .
L'expression (4) se simplifie :
(5)
Là encore : les bornes d'intégration dépendent de la transformation.
3° : la relation (5) est-elle vraiment utile dans la mesure où une transformation réversible : cela n'existe pas ?
La réponse est oui pour deux raisons :
- dans certains cas, assimiler une transformation réelle à une transformation réversible peut constituer une approximation correcte ;
- plus important : comme U et H, S est une fonction d'état ; pour calculer la variation d'entropie lors d'une transformation réelle irréversible,
il est toujours possible d'imaginer un chemin fictif réversible allant de l'état initial réel à l'état final réel.
4° : l'entropie a la dimension physique du quotient d'une énergie par une température ; elle se mesure donc en joules par kelvins (J/K).
5° : comme U et H, S est une fonction d'état extensive : lorsqu'un système est constitué de plusieurs sous-systèmes, l'entropie du système est la somme des entropies des sous-systèmes.
4. Application au thermostat
Un thermostat, en thermodynamique, est un système dont la capacité thermique est tellement grande qu'il peut recevoir ou fournir de la chaleur sans variation mesurable de température.
Imaginons que l'on jette un morceau de métal chaud dans l'eau d'un lac ; le métal va céder de la chaleur à l'eau mais la masse d'eau
est tellement grande devant celle du métal que la variation de température de l'eau sera totalement négligeable.
L'air ambiant constitue aussi un thermostat pour des expériences de durée suffisamment courte pour que l'on puisse négliger les variations de températures en fonction de la météorologie.
On peut aussi envisager bien sûr que le thermostat soit l'air ambiant d'une salle dotée d'un système de régulation de la température, etc.
Soit Tth la température du thermostat et Qth le transfert thermique reçu, au sens algébrique du terme. Le transfert est le plus souvent irréversible. Cependant, irréversible ou réversible, ce transfert ne modifie pas les paramètres d'état du thermostat (température, pression). Puisque l'entropie est une fonction d'état, on peut utiliser la remarque 2 du paragraphe précédent et calculer la variation d'entropie le long d'un chemin fictif réversible isotherme puisque la température du thermostat est fixe. La relation (5) conduit simplement à :
(6)
5. Exercices d'illustration de ce deuxième principe
5.1. Transfert thermique entre un solide et un thermostat
Énoncé
Un solide de capacité thermique constante C est placé dans un four où il acquiert la température initiale T1.
Il est alors sorti du four et laissé à l'air libre. L'air se comporte comme un thermostat de température fixe Text < T1.
1° : Établir les expressions des transferts thermiques reçus par le solide et par l'air en fonction de C, T1 et Text.
2° : Établir l'expression de la variation d'entropie S du solide puis celle de la variation d'entropie Sth du thermostat.
En déduire la variation d'entropie de l'univers : Su. On exprimera cette variation en fonction de C et de .
Le résultat est-il conforme au deuxième principe ?
3° : On s'intéresse maintenant aux cas où l'écart initial de température est très faible en posant : avec .
Effectuer un développement limité de Su au deuxième ordre en fonction de C et .
Commenter le résultat.
Corrigé
1° : L'état final correspond à l'égalité des températures du thermostat et du solide.
Pour le solide : T2 = Text.
Le transfert thermique reçu par le solide vaut :
Il s'agit d'une quantité négative. Le solide cède de la chaleur au thermostat.
L'ensemble « univers » c'est à dire l'ensemble {solide , thermostat} est thermiquement isolé ; il évolue de façon adiabatique :
2° : Pour calculer S nous imaginons une évolution fictive du solide au cours de laquelle il passe réversiblement de la température T1 à la température Text.
Puisque l'état initial et l'état final sont les états réels, on obtient l'expression correcte de la variation d'entropie.
La relation (5) conduit à :
Concernant le thermostat, la relation (6) conduit directement à :
L'entropie étant une fonction extensive :
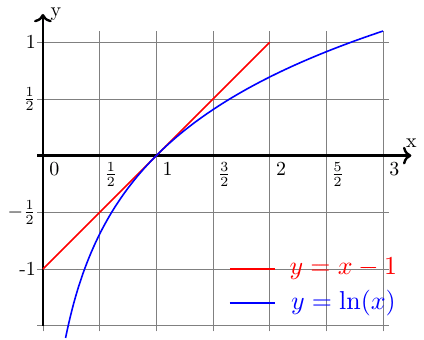
Petit rappel de mathématique illustré figure ci-dessus : la courbe d'équation y = ln(x) admet en x = 1 une tangente oblique d'équation y=(x-1).
On voit clairement que pour x1 :
Ce résultat est conforme au deuxième principe : une évolution adiabatique irréversible se fait avec création d'entropie.
Remarque : on obtient logiquement Su = 0 pour x = 1. Dans ce cas : T1 = Text :
aucun transfert thermique ne se produit ; le système n'évolue pas.
3° : Autre expression de Su:
Développement limité en au voisinage de = 0 :
;
ce qui conduit, pour , à :
On obtient logiquement une augmentation d'entropie d'autant plus importante que l'écart relatif des températures initiales est grand :
plus le déséquilibre thermique initial entre le solide et le thermostat est grand, plus l'entropie créée est importante.
Un peu plus subtil : on peut remarquer que le terme en est nul dans l'expression précédente ;
pour obtenir une entropie créée non nulle, il faut pousser le développement au deuxième ordre.
Cela signifie que, pour des écarts relatifs de température très petits, on peut obtenir une entropie créée nulle au premier ordre près.
Cette situation correspond à un transfert thermique réversible tel que défini précédemment.
5.2. Transfert thermique entre un gaz et un thermostat
Attention : cet exercice comporte quelques subtilités ! Il faut commencer par réfléchir avant d'appliquer des formules ! (cela est toujours vrai mais plus particulièrement ici !)
Énoncé
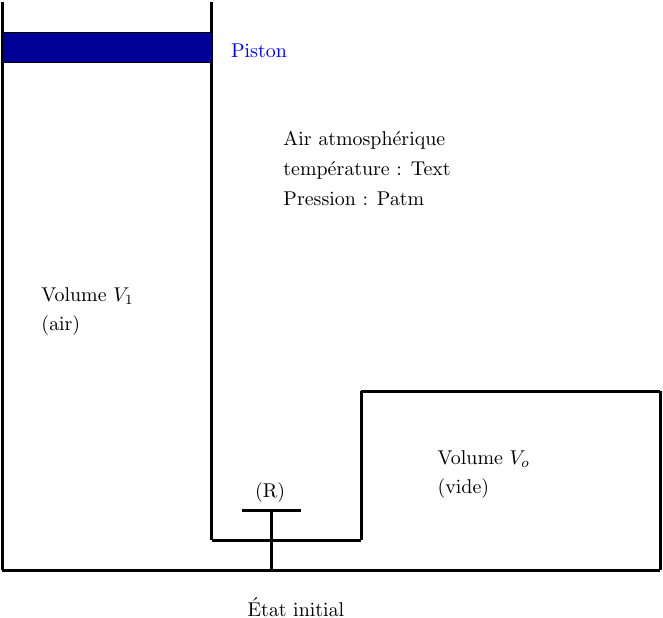
Un réservoir métallique indéformable de volume V0 = 10,0L est vidée de l'air qu'il contenait puis fermé par un robinet (R).
Il est relié à un cylindre métallique d'axe vertical fermé par un piston mobile sans frottement de masse d'influence négligeable.
L'ensemble est placé dans l'air atmosphérique qui se comporte comme un thermostat de température Text = 290 K sous la pression atmosphérique fixe Patm = 0,101 MPa.
Les parois métalliques du cylindre et du réservoir sont de bons conducteurs thermiques (parois diathermanes). L'état initial (état 1) est un état d'équilibre mécanique et thermique de l'air contenu dans le cylindre.
L'air est assimilé à un gaz parfait ; son volume initial est V1 = 20,0 L.
On ouvre alors brusquement le robinet (R) et on attend que l'air retrouve un nouvel état d'équilibre mécanique et thermique : l'état final 2.
1° : Déterminer le travail W reçu par l'air intérieur lors de cette évolution ainsi que le transfert thermique reçu Q.
2° : Déterminer la variation d'entropie S du gaz intérieur puis celle du thermostat Sth puis celle de l'ensemble {air intérieur , air atmosphérique}: Su.
3° : Ce résultat est-il conforme au deuxième principe de la thermodynamique ?
Corrigé
1°: L'état final correspond à la même pression et à la même température que l'état initial.
Puisque la quantité d'air constituant le système étudié est fixe, le volume de gaz est le même qu'à l'état initial :
Attention : l'utilisation sans réfléchir de la relation conduit à W = 0.
Ce résultat est évidemment faux ici puisque le piston exerçant sur le gaz la force F = Patm.S se déplace
(S : aire de la section droite intérieure du cylindre).
En effet, le volume final V2 de gaz est égal au volume initial V1 mais il est réparti différemment :
le volume V0 dans le réservoir et le volume (V1 - V0) dans le cylindre. Le volume de gaz dans le cylindre ayant diminué de V0, le piston s'est enfoncé d'une profondeur .
Le travail de la force F est donc :
Les paramètres d'état (P,V,T) de l'air intérieur étant les mêmes à l'état initial et à l'état final : U2 = U1 puisque l'énergie interne est une fonction d'état.
Cela implique :
Applications numériques :
2° : L'entropie de l'air interne est une fonction d'état comme l'énergie interne.
Le raisonnement fait au-dessus à propos de l'énergie interne s'applique aussi à l'entropie :
L'ensemble {air intérieur , air atmosphérique} est thermiquement isolé :
La relation (6) conduit à :
Application numérique :
L'entropie est une fonction d'état extensive :
3° : Ce résultat est conforme au deuxième principe : une évolution adiabatique irréversible se fait avec création d'entropie.
6. Entropie d'un gaz parfait
On considère n moles d'un gaz parfait dans un état initial 1 caractérisé par les paramètres d'états (P1,T1,V1). Son entropie est S1. Il s'agit d'obtenir l'expression de l'entropie S d'un état d'équilibre quelconque sachant que cet état est caractérisé par les paramètres d'état (P, T, V).
6.1. Les diverses expressions de la variation d'entropie
Ces deux états sont des états d'équilibre, donc des états qui vérifient l'équation d'état.
Des trois paramètres d'état, deux seuls sont indépendants, le troisième étant ensuite déduit de l'équation d'état.
Cela conduit donc à trois expressions différentes selon le couple de paramètres d'état retenu.
Puisque l'entropie est une fonction d'état, la variation d'entropie peut se calculer le long d'un chemin fictif réversible comme déjà expliqué.
6.1.1. Expression en fonction de la température et du volume
Il s'agit d'exprimer la différentielle de l'énergie interne de deux façons différentes sachant que seules les forces de pression sont susceptibles de travailler.
1° : expression en fonction de la variation d'entropie :
(7)
L'évolution est réversible :
(8)
(9)
2° : expression de la première loi de Joule :
(10)
Par identification des deux expressions de dU :
(11)
L'entropie étant une fonction des variables indépendantes T et V, l'expression générale de la différentielle de S s'écrit :
(12)
Par identification :
Intégration :
Attention : la dérivée partielle se calcule à V fixe ;
la « constante » d'intégration peut donc dépendre de V.
Dérivons cette expression par rapport à V en considérant T fixe et identifions avec (11) :
Par intégration :
Cette expression s'applique à l'état initial 1 :
Par soustraction « membre à membre », on fait disparaître la constante :
(13)
ou encore :
(14)
6.1.2. Expression en fonction de la température et de la pression
Elle peut se déduire de (14) en tenant compte de l'équation d'état des gaz parfaits.
On peut aussi utiliser la méthode du paragraphe précédent en remplaçant dU par dH. Les justificatifs des calculs sont les mêmes.
(15)
Deuxième loi de Joule :
(16)
(17)
(18)
Par identification et intégration :
Par intégration :
Cas particulier de l'état 1 :
Soustraction membre à membre :
(19)
Soit aussi :
(20)
6.1.3. Expression en fonction de la pression et du volume
La méthode la plus simple consiste à partir de l'expression (14) en tenant compte de l'équation d'état des gaz parfaits :
Selon (14) :
(21)
6.1.4. Retour sur la loi de Laplace
Cette loi a été étudiée au paragraphe 6 de la fiche sur le premier principe.
On rappelle les conditions de validité de cette loi :
1° : seules les forces de pression sont susceptibles de travailler ;
2° : évolution quasi statique ;
3° : gaz assimilé à un gaz parfait ;
4° : évolution adiabatique : aucun transfert thermique n'intervient lors de l'évolution, ce qui suppose les parois du cylindre et du piston athermanes, c'est à dire suffisamment bien isolées thermiquement du milieu extérieur.
On remarque que considérer la transformation comme isentropique (évolution à entropie constante) permet de retrouver les trois expressions équivalentes de la loi de Laplace.
En écrivant S = S1 pour tout état du gaz parfait, on obtient en effet :
(22)
Un gaz parfait est parfaitement élastique.
Une transformation quasi statique d'un tel gaz est donc aussi renversable ; elle est ainsi réversible.
Or, selon le deuxième principe, une transformation adiabatique et réversible est isentropique.
6.2. Entropie de mélange de deux gaz parfaits
On envisage deux réservoirs aux parois diathermanes, reliés par un tube muni d'un robinet initialement fermé. Le premier réservoir contient n1 moles d'un gaz parfait n° 1 sous la pression P0 à la température T0 = Text. Le second réservoir contient n2 moles d'un gaz parfait n° 2 sous la même pression P0 à la même température T0=Text. On ouvre alors le robinet et on attend que le mélange devienne homogène. Il s'agit de déterminer la variation d'entropie du système formé des deux gaz lors de leur mélange. On suppose la température de l'air extérieure fixe. Le gaz n°1, le gaz n°2 et le mélange sont des gaz parfaits. L'équation d'état appliquée aux trois gaz conduit à :
Le mélange final occupe le volume (V1+V2) à la même température Text :
(23)
La pression n'est pas modifiée par le mélange à température fixe.
6.2.1. Cas simple où les deux gaz sont identiques
Le cas est trivial : l'agitation thermique fait qu'un certain nombre de molécules du réservoir n°1 passent dans le réservoir n° 2 et inversement, les quantités de molécules dans chaque réservoir restant en moyenne constante.
Les molécules des deux gaz étant indiscernables, l'état final et l'état initial sont strictement identiques.
L'entropie étant une fonction d'état, le mélange ne provoque aucune variation d'entropie :
6.2.2. Notion de pression partielle ; loi de Dalton
On commence par définir la notion de pression partielle d'un gaz appartenant à un mélange de N gaz parfaits. On définit d'abord la fraction molaire du gaz n° i, c'est à dire la proportion molaire de ce gaz dans le mélange :
(25)
Bien sûr, il résulte de cette définition :
(26)
Si P désigne la pression totale du mélange, la pression partielle du gaz n°i est :
(27)
Compte tenu de (26) :
(28)
En notant V le volume du mélange gazeux et T sa température, l'équation d'état conduit à :
(29)
Selon (25), (27) et (29) :
(30)
On voit ainsi que la pression partielle du gaz n°i est la pression qu'aurait le gaz n°i s'il occupait seul la totalité du volume V à la température T du mélange. Ce résultat est connu sous le nom de loi de Dalton. Un peu plus généralement, on peut dire que chaque gaz du mélange de gaz parfait a les même propriétés thermodynamiques que s'il occupait seul la totalité du volume V à la température T du mélange sous une pression égale à sa pression partielle.
6.2.3. Variation d'entropie lors du mélange de deux gaz parfaits différents
On revient à l'expérience décrite en début de paragraphe 6.2.
Compte tenu de ce qui vient d'être dit, la variation d'entropie du gaz n° 1 se calcule en supposant que ce gaz passe, à température fixe, de la pression P0 à la pression finale égale à sa pression partielle finale :
(31)
La variation d'entropie de ce gaz n°1 se déduit de (19) :
(32)
Le raisonnement est identique pour le gaz n°2 : il suffit de permuter les indices 1 et 2 :
(33)
L'entropie étant une fonction extensive, l'expression de la variation d'entropie lors du mélange s'écrit :
(34)
Cette variation est évidemment strictement positive. Les températures initiale et finale étant égales, la première loi de Joule conduit à
Puisque toutes les parois sont fixes (pas de piston mobile) : W = 0 ; donc : Q = 0.
Le mélange s'effectue de façon adiabatique. Le mélange est une transformation non renversable :
on ne peut imaginer que les molécule n°1 retournent spontanément dans le réservoir n°1 et les molécules n°2 dans le réservoir n°2 !
Puisque l'évolution est adiabatique irréversible, elle se produit avec augmentation de l'entropie.
7. Entropie et désordre à l'échelle microscopique
7.1. Généralités
L'étude rigoureuse de ce lien relève de la thermodynamique statistique.
On se contente ici de quelques notions qualitatives sans démonstration.
On va donc admettre que l'entropie d'un système est d'autant plus élevée que le désordre à l'échelle microscopique est élevé.
L'étude précédente de la variation d'entropie lors d'un mélange de deux gaz parfaits en est une bonne illustration :
le désordre à l'état final y est évidemment plus important qu'à l'état initial.
Revenons maintenant au cas d'un gaz parfait pur.
La relation (14) montre que l'entropie d'un gaz parfait est une fonction croissante de sa température et une fonction croissante de son volume.
Augmenter le volume d'un gaz augmente l'espace où les molécules sont susceptibles de se déplacer ; augmenter la température augmente l'agitation thermique ; ces deux augmentations contribuent à augmenter le désordre à l'échelle moléculaire.
De façon plus générale, une augmentation de température augmente l'agitation thermique donc augmente le désordre à l'échelle macroscopique.
On peut donc considérer que l'entropie d'un système quelconque est une fonction croissante de la température.
7.2. Interprétation de l'expérience décrite au paragraphe 1.
Le système est l'eau enfermé dans un calorimètre supposé parfait. L'évolution est nécessairement adiabatique ; on envisage une évolution spontanée donc irréversible ; cela conduit à deux équations :
(35)
Premier cas : W > 0 : le système reçoit du travail ; cela augmente l'énergie interne donc augmente l'agitation thermique et le désordre à l'échelle microscopique.
Cela est tout à fait en accord avec une augmentation d'entropie. Cette transformation est possible. Deuxième cas : W < 0 : une fois le moteur enlevé, on pourrait envisager que le liquide, en se refroidissant, perde de l'énergie interne en faisant tourner les pales.
Ce refroidissement adiabatique du liquide entraînerait une diminution d'entropie interdite par le deuxième principe.
Cette évolution est donc impossible.
8. Application des deux principes à l'étude de cycles thermodynamiques
8.1. Généralités
Le système étudié est un système fermé constitué d'un fluide, parfois un gaz, parfois un corps pur pouvant être présent sous deux phases : une phase vapeur et une phase liquide, comme dans les centrales thermiques, les réfrigérateurs ou les pompes à chaleur. L'évolution est cyclique : le fluide retrouve à intervalles de temps fixes le même état, la succession d'états intermédiaires entre ces deux états identiques étant toujours la même. Ce genre d'évolution se rencontre dans diverses machines comme certains moteurs tournant à vitesse constante, les moteurs à vapeur, les réfrigérateurs, les pompes à chaleur, etc..., etc. Dans un diagramme de Clapeyron représentant les variations de la pression en fonction du volume, la représentation des divers états successifs correspond à une courbe fermée.
Par la suite, toutes les variations et toutes les quantités prises en compte correspondront à un cycle.
Propriété
Les variations de fonction d'état sur un cycle sont donc évidemment nulle puisqu'elles sont calculées entre un état initial et un état final identiques :
(36)
(37)
8.2. Cas d'un cycle monotherme
Le transfert thermique au cours du cycle ne peut s'effectuer qu'avec une seule source de température fixe Ts.
Cette source est donc assimilable à un thermostat.
La variation d'entropie s'obtient à partir de (37) et (4) :
(38)
Puisque : :
(39)
Selon (36) :
(40)
En tenant compte de (39) et (40) :
(41)
Ainsi : un cycle monotherme ne peut en aucun cas être moteur c'est à dire fournir du travail à l'extérieur.
Il n'est pas possible de fabriquer un moteur thermique à partir d'une seule source de chaleur.
8.3. Cas d'un cycle ditherme
8.3.1. Généralités
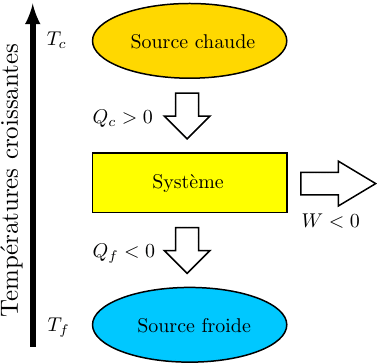
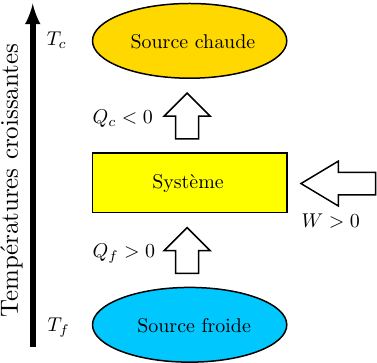
Au cours du cycle, on obtient deux transferts thermiques : un transfert Qc avec une source chaude équivalente à un thermostat de température Tc et un transfert avec une source froide équivalente à un thermostat de température Tf < Tc. On note W le travail reçu (au sens algébrique du terme) par le système sur un cycle.
Le premier principe de la thermodynamique conduit à :
(42)
Le deuxième principe de la thermodynamique conduit à :
(43)
Remarque : la relation (43) est équivalente à :
(44)
La suite de l'étude peut se faire en raisonnant sur cette inégalité connue sous le nom d'inégalité de Clausius.
On conseille cependant aux étudiants peu à l'aise avec le maniement mathématique des inégalités, surtout si elles concernent des quantités négatives, à plutôt utiliser l'égalité (43) ; la conclusion finale s'obtenant en remarquant : .
8.3.2. Cycle ditherme moteur
Il s'agit d'exploiter le fait qu'un transfert thermique se fait spontanément d'un milieu chaud vers un milieu froid. En effectuant ce transfert par l'intermédiaire du fluide décrivant le cycle (le système thermodynamique) il est possible de récupérer du travail. Les sens des transferts et les signes des différentes quantités sont visualisés sur la figure ci-dessus. Il peuvent être démontrés à partir des relations ci-dessus :
selon (42), si W < 0 :
(45)
Puisque les deux températures sont strictement positives, cette inégalité (45) n'est compatibles avec l'inégalité (44) que si les deux transferts thermiques sont de signes différents.
(46)
Selon (43) :
(47)
Selon (42) et (47) :
On remarque : et ;
cela conduit à : Qc > 0 et , compte tenu (46) : Qf < 0.
Ce résultat est connu sous le nom de théorème de Carnot dont un des énoncés possibles est le suivant :
Théorème de Carnot
Pour qu'un cycle ditherme soit moteur, le système décrivant le cycle doit absorber de la chaleur à la source chaude et en rejeter à la source froide.
Efficacité thermodynamique du cycle :
Définition
L'efficacité d'une machine a pour définition générale :
(48)
La quantité utile pour un moteur est évidemment le travail fourni à l'extérieur soit |W| = -W ;
la source chaude correspond, dans les centrales électriques par exemple, à une chaudière qu'il faut maintenir à température élevée grâce à un combustible (gaz, charbon, combustible nucléaire, etc.) qu'il faut payer.
En revanche, la source froide est l'eau d'une rivière passant au voisinage de la centrale ou l'air ambiant.
Si le réchauffement de l'air ambiant ou de l'eau des rivières peut avoir des conséquences écologiques, ce rejet d'énergie thermique à la source froide n'est pas coûteux.
La quantité coûteuse est donc Qc.
Donc, pour un cycle moteur ditherme :
(49)
Soit en tenant compte de (42) :
(50)
(47) :
(51)
(52)
; cela conduit à :
(53)
Dans le cas d'un moteur, l'efficacité peut aussi être appelé « rendement ».
La valeur maximale de ce rendement, souvent appelée rendement de Carnot, représente le rendement dans le cas limite d'une évolution réversible.
On peut remarquer que ce rendement, toujours inférieur à l'unité, est d'autant plus élevé que l'écart de températures entre source chaude et source froide est important.
8.3.3. Cycles dithermes récepteurs
Il s'agit d'inverser , par rapport au cas précédent, le sens du transfert thermique en faisant passer de la chaleur de la source froide vers la source chaude par l'intermédiaire du fluide décrivant le cycle.
Ce transfert n'est pas naturel : pour qu'il soit possible, le système doit recevoir de l'énergie mécanique de l'extérieur : W > 0 ;
d'où les signes précisés sur la figure ci-dessus. Suivant l'usage que l'on fait de la machine, on peut distinguer deux cas.
a) les frigopompes (réfrigérateurs, congélateurs, climatiseurs, etc.) : il s'agit de maintenir une enceinte (intérieur du réfrigérateur ou du congélateur, salle d'habitation, etc.) plus froide que le milieu extérieur. Puisque cette enceinte n'est pas parfaitement isolée de l'extérieur, il est nécessaire de prélever à l'intérieur de cette enceinte qui constitue la source froide, de la chaleur qu'il faut rejeter dans le milieu extérieur qui constitue la source chaude. La quantité utile est donc Qf et la quantité coûteuse est évidement le travail : le compresseur de la frigopompe est entraîné par un moteur électrique, etc. L'efficacité de la frigopompe est ainsi :
(54)
en tenant compte de (43) :
(55)
(56)
Puisque : :
(57)
On remarque que l'efficacité maximale correspond au cas limite de la réversibilité.
L'efficacité est, dans les situations usuelles, supérieure à l'unité ;
elle est d'autant plus grande que l'écart de température entre source chaude et source froide est faible.
b) les thermopompes (ou pompes à chaleur) : il s'agit de maintenir l'hiver une enceinte (souvent l'intérieur d'une habitation) à une température supérieure à la température extérieure.
Comme cette enceinte, qui constitue la source chaude) n'est pas parfaitement isolée thermiquement, il faut prélever au milieu extérieur, qui constitue la source froide de la chaleur pour en fournir à l'intérieur de l'habitation qui constitue la source chaude.
La quantité utile est donc ici . Comme dans le cas précédent, la quantité coûteuse est W.
D'où l'expression de l'efficacité de la thermopompe :
(58)
En tenant compte de (51) :
Puisque : et :
(59)
L'efficacité maximale correspond au cas limite de la réversibilité. Elle est strictement supérieure à l'unité. On remarque aussi qu'elle est d'autant plus grande que l'écart de température entre les deux sources est faible. L'efficacité des thermopompes est aussi appelée « coefficient de performance » : abréviation anglo-saxonne consacrée par l'usage : « COP ».
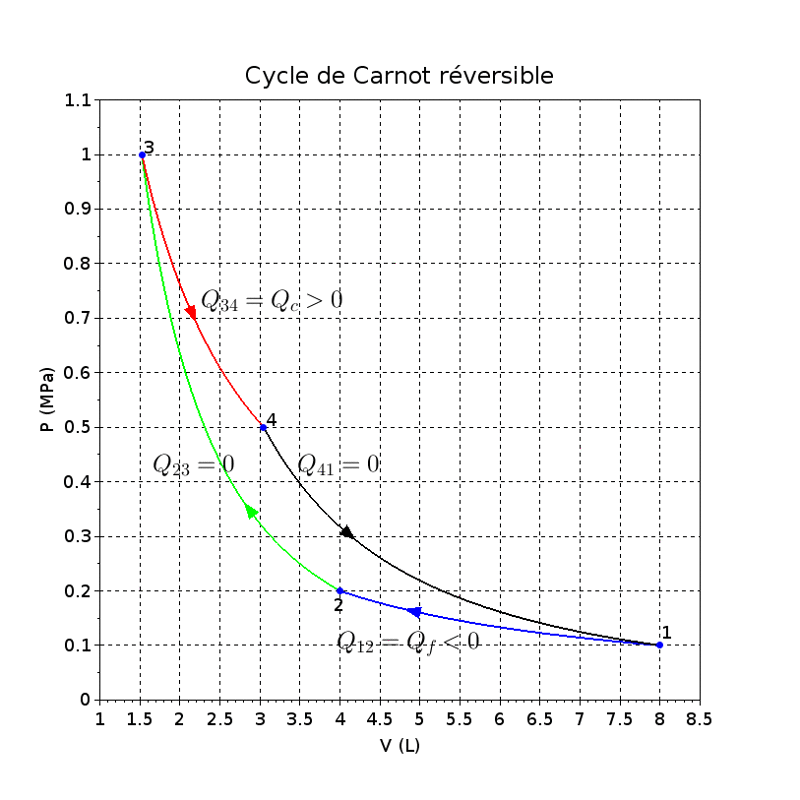
9. Le cycle de Carnot moteur
9.1. Généralités
Le fluide décrivant le cycle est assimilé à un gaz parfait monoatomique et le cycle est supposé décrit réversiblement.
Il est constitué de deux évolutions adiabatiques alternant avec deux évolutions isothermes conformément à la figure ci-dessus ci-contre :
- évolution 1-2 : le gaz parfait subit une compression isotherme à la température Tf = 290K au cours de laquelle il rejette à l'extérieur de la chaleur ;
- évolution 2-3 : le gaz subit une compression adiabatique au cours de laquelle il s'échauffe jusqu'à la température T3 = Tc = 552K ;
- évolution 3-4 : le gaz subit une détente isotherme à la température Tc au cours de laquelle il reçoit de la chaleur de l'extérieur ;
- évolution 4-1 : le gaz subit une détente adiabatique au cours de laquelle il se refroidit jusqu'à la température T1 = Tf.
On peut justifier l'allure du cycle en comparant les coefficient directeurs de l'adiabatique et de l'isotherme en un même point de coordonnées (V,P) dans le diagramme de Clapeyron (points 2 et 4).
Le long d'une isotherme pour un gaz parfait évoluant de façon réversible :
(60)
En différenciant :
(61)
D'où l'expression générale du coefficient directeur de la tangente à une isotherme dans le diagramme de Clapeyron :
(62)
Le long de l'isentropique :
En différenciant :
(63)
D'où l'expression générale du coefficient directeur de la tangente à une isentropique dans le diagramme de Clapeyron :
(64)
Dans le diagramme de Clapeyron, en un même point de coordonnées (V,P) :
(65)
Cela explique les ruptures de pentes dans le diagramme de Clapeyron du cycle de Carnot aux points 2 et 4.
La tangente en ces points à la courbe isentropique est fois plus « pentue » que la tangente à la courbe isotherme.
La valeur de est 1,67 pour les gaz monoatomiques et 1,40 pour les gaz diatomiques.
La rupture de pente est donc relativement faible, d'où la forme très « effilée » du cycle de Carnot.
Rien à voir avec certaines représentations du cycle de Carnot où, pour mieux différentier les isothermes des isentropiques, on accentue fortement l'écart de pente.
Il est aussi possible de déterminer graphiquement le travail fourni à partir du cycle.
(66)
où A1 désigne l'aire de la surface sous la courbe 3-2-1 (A1>0).
(67)
où A2 désigne l'aire de la surface sous la courbe 3-4-1 (A2>0).
Le travail reçue par le fluide constituant le système sur un cycle vaut :
Pour un cycle moteur, ce qui correspond à un cycle décrit dans le sens de rotation des aiguilles d'une montre,
le travail est négatif et sa valeur absolue est égale à l'aire de la surface du cycle dans le diagramme de Clapeyron.
9.2. Impossibilité pratique du cycle de Carnot
Ce cycle est la source quasi inépuisable d'exercices au niveau bac+1. Il est cependant impossible de le mettre en oeuvre, même de façon approchée, à l'échelle industrielle.
9.2.1. Cas où le fluide décrivant le cycle est un gaz
Une puissance du moteur importante suppose que le cycle soit décrit rapidement ; le nombre de cycles par minute doit être élevé.
Impossible dans ses conditions d'avoir des transformations rigoureusement quasi statiques.
Nous allons montrer qu'il est impossible de réaliser le cycle même de façon approchée.
Réaliser une transformation adiabatique est assez simple.
On sait que les transferts thermiques se font toujours lentement ;
une transformation rapide, même avec des parois métalliques conductrices de la chaleur est assimilable à une transformation adiabatique.
En revanche, une compression ou une détente isotherme doit toujours se produire lentement pour laisser le temps au transfert thermique de se produire à travers la paroi du cylindre.
On voit bien qu'alterner, dans une même machine, des évolution adiabatiques rapides et des évolutions isothermes lentes est incompatible avec une rotation régulière du moteur.
9.2.2. Cas où le fluide décrivant le cycle est en équilibre liquide-vapeur
Cette situation se rencontre essentiellement dans les centrales thermiques (cycles moteurs) et dans les thermopompes et frigopompes (cycles récepteurs). On sait que la température d'ébullition d'un liquide reste fixe pendant toute l'ébullition si la pression est maintenue fixe.
Plus généralement, un corps pur en équilibre liquide-vapeur garde une température fixe tant que la pression reste fixe.
Il est donc aisé d'obtenir ainsi des évolutions isothermes dans la mesure où il est facile techniquement de maintenir la pression fixe. Cela règle le problème des évolutions 1-2 et 3-4. La détente 4-1 du mélange peut être rendue adiabatique pour peu qu'elle soit rapide.
Reste le problème de la compression adiabatique 2-3 : on ne sait pas fabriquer de compresseur pour les mélanges liquide-vapeur dans la mesure où un liquide ne diminue quasiment pas de volume lors d'une compression alors que la vapeur diminue fortement de volume par compression.
10. Le cycle moteur de Stirling
10.1. Étude théorique du cycle réversible
Le système est constitué des n moles de gaz parfait décrivant le cycle composé de quatre transformations avec Tc = 700 K et Tf = 350 K :
- Transformation 1-2 : compression isotherme à la température Tf :
(68)
Première loi de Joule : à T fixe, l'énergie interne reste fixe :
(69)
- Transformation 2-3 : compression isochore :
(70)
- Transformation 3-4 : détente isotherme à la température Tc : l'étude est analogue à celle de la transformation 1-2 :
(71)
- Transformation 4-1 : détente isochore : l'étude est analogue à celle de la transformation 2-3 :
(72)
Le cycle de Stirling, tel que décrit pour l'instant, n'est pas ditherme dans la mesure où les transformations 2-3 et 4-1 correspondent à des transferts thermiques non nul. C'est là qu'intervient le rôle du récupérateur : il s'agit d'un dispositif qui absorbe la chaleur perdue par le système lors de l'étape 4-1 pour la réinjecter dans le système lors de l'étape 2-3. Cela ne modifie pas le cycle puisque nous avons montré : ; cela permet d'assimiler le cycle à un cycle ditherme réversible. Vérifions qu'il est possible de retrouver les propriétés générales établies précédemment pour les cycles dithermes réversibles.
Les relations (69) et (71) permettent d'écrire :
(73)
On retrouve l'égalité de Clausius (44) dans le cas particulier de la réversibilité.
Le travail total reçue par le gaz décrivant le cycle, quantité calculée sur un cycle, vaut :
(74)
La quantité coûteuse Qc est fournie par (71). Le rendement du cycle dans le cas de la réversibilité vaut ainsi :
(75)
On retrouve bien l'expression du rendement de Carnot (voir 59), qui, dans ce cas particulier vaut 50%.
10.2. Intérêt pratique du moteur Stirling.
Le rendement réel est évidemment inférieur au précédent pour plusieurs raisons :
- Pour obtenir une puissance non négligeable du moteur, la durée de chaque cycle doit être courte ;
les évolutions doivent être rapides donc non quasi statiques.
- Les évolution considérées comme isothermes ne le sont pas tout à fait malgré la présence d'ailettes sur les cylindres qui augmente la vitesse des transferts thermiques.
- L'efficacité du récupérateur n'est pas de 100% : la chaleur récupérée lors de l'étape 4-1 n'est pas intégralement restituée lors de l'étape 2-3.
Le moteur est aussi assez coûteux à fabriquer, en particulier à cause des étapes isochores.
Il est évidemment impossible d'obtenir de telles transformations avec un seul cylindre fermé par un piston en mouvement de va et vient permanent.
Diverses astuces peuvent être utilisées pour obtenir, du moins de façon approchée, de telles transformations isochores : l'utilisation de deux cylindres reliés à un même arbre, par exemple.
Cependant, ce moteur a trouvé un regain d'intérêt car, le côté chaud du moteur placé au voisinage du foyer d'un miroir parabolique, ce moteur, associé à un alternateur, permet de convertir l'énergie solaire en énergie électrique avec le meilleur taux de conversion actuellement observé :
un peu plus de 30% alors que les panneaux photovoltaïques en sont à 20% environ.
Publié par gbm / relue par gbm
le
ceci n'est qu'un extrait
Pour visualiser la totalité des cours vous devez vous inscrire / connecter (GRATUIT) Inscription Gratuitese connecter
Merci à vanoise pour avoir contribué à l'élaboration de cette fiche
Désolé, votre version d'Internet Explorer est plus que périmée ! Merci de le mettre à jour ou de télécharger Firefox ou Google Chrome pour utiliser le site. Votre ordinateur vous remerciera !
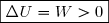
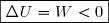
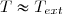 }.
Si le système est séparé de l'extérieur par une parois mobile (piston dans un cylindre par exemple), on doit avoir à chaque instant :
}.
Si le système est séparé de l'extérieur par une parois mobile (piston dans un cylindre par exemple), on doit avoir à chaque instant : 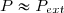 .
.
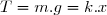 où
où 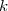 est la constante de raideur du ressort. Si on enlève progressivement et lentement les masselottes, le ressort va retrouver son état initial correspondant au point O, tous les états intermédiaires étant encore sur le segment de droite rouge. Bref : le ressort revient à son état initial par la même succession d'états intermédiaires. La transformation est donc renversable. Puisqu'elle est aussi quasi statique, elle est réversible.
est la constante de raideur du ressort. Si on enlève progressivement et lentement les masselottes, le ressort va retrouver son état initial correspondant au point O, tous les états intermédiaires étant encore sur le segment de droite rouge. Bref : le ressort revient à son état initial par la même succession d'états intermédiaires. La transformation est donc renversable. Puisqu'elle est aussi quasi statique, elle est réversible.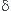 W, un transfert thermique
W, un transfert thermique 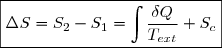 (4)
(4)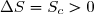 .
.
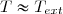 .
L'expression (4) se simplifie :
.
L'expression (4) se simplifie :
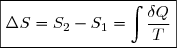 (5)
(5)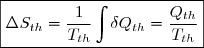 (6)
(6) S du solide puis celle de la variation d'entropie
S du solide puis celle de la variation d'entropie 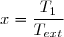 .
Le résultat est-il conforme au deuxième principe ?
.
Le résultat est-il conforme au deuxième principe ?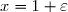 avec
avec 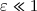 .
Effectuer un développement limité de
.
Effectuer un développement limité de 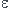 .
Commenter le résultat.
.
Commenter le résultat.)
)
)
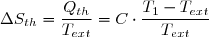
![\Delta S_{u} = \Delta S + \Delta S_{th} = C \left[x-1- \ln \left(x \right) \right]](https://latex.ilephysique.net/latex-0.tex?\Delta S_{u} = \Delta S + \Delta S_{th} = C \left[x-1- \ln \left(x \right) \right])
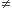 1 :
1 :\quad \text{soit : \ensuremath{ \quad \Delta S_{u} > 0}})
![\Delta S_{u} = C \cdot \left[\varepsilon - \ln \left(1+ \varepsilon \right) \right]](https://latex.ilephysique.net/latex-0.tex?\Delta S_{u} = C \cdot \left[\varepsilon - \ln \left(1+ \varepsilon \right) \right])
 = \varepsilon - \dfrac{1}{2} \varepsilon^{2} + o \left( \varepsilon^{3} \right)) ;
ce qui conduit, pour
;
ce qui conduit, pour 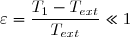 , à :
, à :
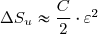
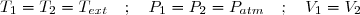
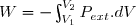 conduit à W = 0.
Ce résultat est évidemment faux ici puisque le piston exerçant sur le gaz la force F = Patm.S se déplace
(S : aire de la section droite intérieure du cylindre).
En effet, le volume final V2 de gaz est égal au volume initial V1 mais il est réparti différemment :
le volume V0 dans le réservoir et le volume (V1 - V0) dans le cylindre. Le volume de gaz dans le cylindre ayant diminué de V0, le piston s'est enfoncé d'une profondeur
conduit à W = 0.
Ce résultat est évidemment faux ici puisque le piston exerçant sur le gaz la force F = Patm.S se déplace
(S : aire de la section droite intérieure du cylindre).
En effet, le volume final V2 de gaz est égal au volume initial V1 mais il est réparti différemment :
le volume V0 dans le réservoir et le volume (V1 - V0) dans le cylindre. Le volume de gaz dans le cylindre ayant diminué de V0, le piston s'est enfoncé d'une profondeur 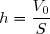 .
Le travail de la force F est donc :
.
Le travail de la force F est donc :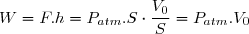
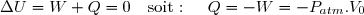
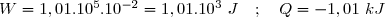
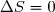
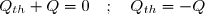
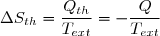
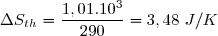
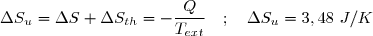
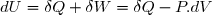 (7)
(7)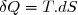 (8)
(8)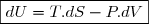 (9)
(9)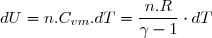 (10)
(10)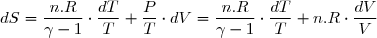 (11)
(11)_{V} \cdot dT+ \left(\dfrac{\partial S}{\partial V}\right)_{T}\cdot dV) (12)
(12)_{V} = \dfrac{n.R}{\gamma-1} \cdot \dfrac{1}{T})
 + f(V))
_{V}) se calcule à V fixe ;
la « constante » d'intégration peut donc dépendre de V.
Dérivons cette expression par rapport à V en considérant T fixe et identifions avec (11) :
se calcule à V fixe ;
la « constante » d'intégration peut donc dépendre de V.
Dérivons cette expression par rapport à V en considérant T fixe et identifions avec (11) :_{T} = f'(V) = \dfrac{n.R}{V})
 = n . R . \ln \left(V \right)+K \qquad \text{avec K : constante})
+ n . R . \ln \left(V \right) + K)
 + n . R . \ln \left(V_{1}\right) + K)
+ n . R . \ln \left(\dfrac{V}{V_{1}} \right)) (13)
(13)}) (14)
(14)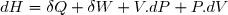
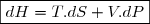 (15)
(15)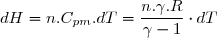 (16)
(16)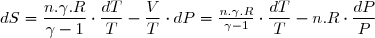 (17)
(17)_{P} \cdot dT + \left(\dfrac{\partial S}{\partial P} \right)_{T} \cdot dP) (18)
(18)_{P} = \dfrac{n . \gamma . R}{\gamma - 1} \cdot \dfrac{1}{T} \quad ; \quad S = \dfrac{n . \gamma . R}{\gamma - 1} \cdot \ln \left(T \right) + g(P))
_{T} = - \dfrac{n . R}{P} = g'(P))
 = - n . R . \ln \left(P \right) + K' \qquad \text{avec K' : constante})
- n . R . \ln \left(P \right) + K')
 - n . R . \ln \left(P_{1} \right) + K')
 - n . R . \ln \left(\dfrac{P}{P_{1}} \right)) (19)
(19)![\boxed{S - S_{1} = \dfrac{n . \gamma . R}{\gamma - 1} \cdot \ln \left[ \left(\dfrac{T}{T_{1}} \right) \cdot \left(\dfrac{P_{1}}{P}\right)^{\dfrac{\gamma-1}{\gamma}} \right]}](https://latex.ilephysique.net/latex-0.tex?\boxed{S - S_{1} = \dfrac{n . \gamma . R}{\gamma - 1} \cdot \ln \left[ \left(\dfrac{T}{T_{1}} \right) \cdot \left(\dfrac{P_{1}}{P}\right)^{\dfrac{\gamma-1}{\gamma}} \right]}) (20)
(20)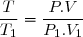
)
}) (21)
(21)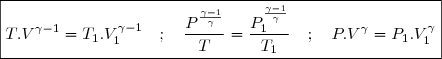 (22)
(22)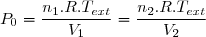
 . R . T_{ext}}{V_{1} + V_{2}} = \dfrac{R . T_{ext}}{V_{1} + V_{2}} \cdot \left(\dfrac{P_{0} . V_{1}}{R . T_{ext}} + \dfrac{P_{0} . V_{2}}{R . T_{ext}} \right) = P_{0}) (23)
(23)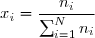 (25)
(25)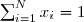 (26)
(26)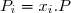 (27)
(27)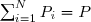 (28)
(28)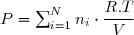 (29)
(29)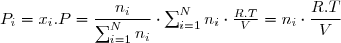 (30)
(30)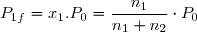 (31)
(31) = n_{1} R . \ln \left(\dfrac{n_{1} + n_{2}}{n_{1}} \right)) (32)
(32)) (33)
(33) + n_{2}R . \ln \left(\dfrac{n_{1} + n_{2}}{n_{2}} \right)}) (34)
(34)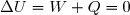
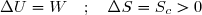 (35)
(35)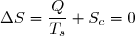 (38)
(38)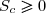 :
: (39)
(39) (41)
(41)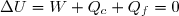 (42)
(42)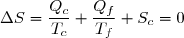 (43)
(43)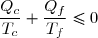 (44)
(44) .
.
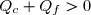 (45)
(45)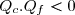 (46)
(46)) (47)
(47)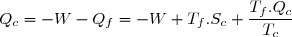
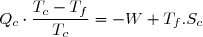
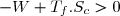 et
et 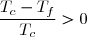 ;
cela conduit à : Qc > 0 et , compte tenu (46) : Qf < 0.
Ce résultat est connu sous le nom de théorème de Carnot dont un des énoncés possibles est le suivant :
;
cela conduit à : Qc > 0 et , compte tenu (46) : Qf < 0.
Ce résultat est connu sous le nom de théorème de Carnot dont un des énoncés possibles est le suivant :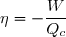 (49)
(49)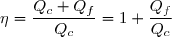 (50)
(50)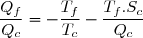 (51)
(51)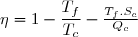 (52)
(52) ; cela conduit à :
; cela conduit à : (53)
(53)
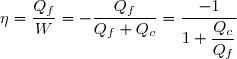 (54)
(54)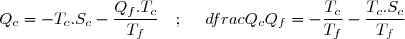 (55)
(55)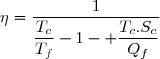 (56)
(56)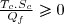 :
: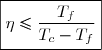 (57)
(57)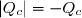 . Comme dans le cas précédent, la quantité coûteuse est W.
D'où l'expression de l'efficacité de la thermopompe :
. Comme dans le cas précédent, la quantité coûteuse est W.
D'où l'expression de l'efficacité de la thermopompe :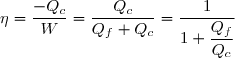 (58)
(58)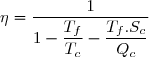
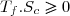 et
et 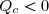 :
: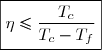 (59)
(59)
) et le cycle est supposé décrit réversiblement.
Il est constitué de deux évolutions adiabatiques alternant avec deux évolutions isothermes conformément à la figure ci-dessus ci-contre :
et le cycle est supposé décrit réversiblement.
Il est constitué de deux évolutions adiabatiques alternant avec deux évolutions isothermes conformément à la figure ci-dessus ci-contre : + \ln \left(V \right) = \ln \left(K \right)) (60)
(60)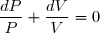 (61)
(61)_{T} = - \dfrac{P}{V}}) (62)
(62) + \gamma . \ln \left(V \right) = \ln \left(K' \right))
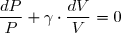 (63)
(63)_{S} = -\gamma \cdot \dfrac{P}{V}}) (64)
(64)_{S} = \gamma \cdot \left(\dfrac{\partial P}{\partial V} \right)_{T}) (65)
(65)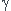 fois plus « pentue » que la tangente à la courbe isotherme.
La valeur de
fois plus « pentue » que la tangente à la courbe isotherme.
La valeur de 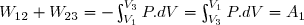 (66)
(66)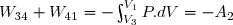 (67)
(67)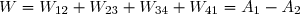
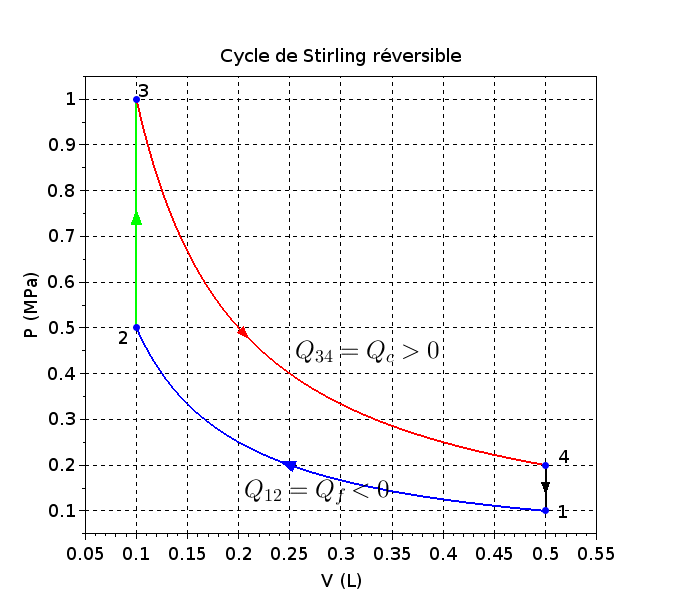
) (68)
(68)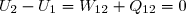
) (69)
(69)) (70)
(70) \quad ; \quad Q_{34} = Q_{c} = n . R . T_{c} . \ln \left(\dfrac{V_{1}}{V_{2}} \right)) (71)
(71)) (72)
(72)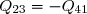 ; cela permet d'assimiler le cycle à un cycle ditherme réversible. Vérifions qu'il est possible de retrouver les propriétés générales établies précédemment pour les cycles dithermes réversibles.
; cela permet d'assimiler le cycle à un cycle ditherme réversible. Vérifions qu'il est possible de retrouver les propriétés générales établies précédemment pour les cycles dithermes réversibles. - n . R . \ln \left(\dfrac{V_{1}}{V_{2}}\right) = 0) (73)
(73) . n . R .\ln \left(\dfrac{V_{1}}{V_{2}} \right)) (74)
(74)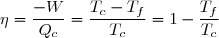 (75)
(75)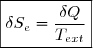 (1)
(1)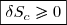 (2)
(2)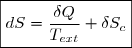 (3)
(3)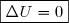 (36)
(36)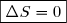 (37)
(37) (48)
(48) forums de physique - chimie
forums de physique - chimie