Cette fiche concerne la thermodynamique dite « macroscopique ».
L'état du système étudié est caractérisé par un certain nombre de paramètres tel que la pression, la température, le volume, la masse volumique, etc.
La thermodynamique « microscopique » ou « statistique » où on s'intéresse aux mouvement des différentes molécules du système n'est pas étudiée ici.
Cette fiche se limite à l'étude des systèmes n'échangeant pas de matière avec le milieu extérieur, c'est à dire à l'étude des systèmes fermés, et se termine par un exercice d'illustration des notions présentées.
1. Objectif
Il s'agit de généraliser le théorème de l'énergie cinétique appliqué à un système fermé.
Théorème de l'énergie cinétique
La variation d'énergie cinétique entre un instant initial (indice 1) et un instant final (indice 2) est égal au travail de toutes les forces appliquées au système :
(1)
Il s'agit ici de généraliser ce théorème en prenant en compte deux termes supplémentaires :
1° : on tient compte dorénavant des transferts thermiques : la variation d'énergie tient compte dorénavant des échanges de quantités de chaleur .
2° : on tient compte de l'énergie interne, notée , du système.
Cette énergie interne est une somme de deux termes :
*l'énergie cinétique d'agitation thermique : il s'agit de la somme des énergies cinétiques des différentes particules constituant le système étudié, les vitesses étant les vitesses mesurées dans le repère barycentrique du système. Il est important de bien distinguer cette énergie cinétique liée au mouvement désordonné des particules à l'énergie cinétique macroscopique qui s'intéresse au mouvement d'ensemble des particules par rapport au repère d'étude.
Exemple : un fluide qui s'écoule dans une canalisation à une certaine vitesse possède à la fois une énergie cinétique due à l'écoulement et une énergie cinétique d'agitation thermique prise en compte dans la valeur de l'énergie interne .
*l'énergie potentielle d'interaction entre les différentes particules du système. Elle est essentiellement d'origine électrique (forces de Van der Waals par exemple).
Généralisation du théorème de l'énergie cinétique
La généralisation du théorème de l'énergie cinétique s'écrit donc finalement :
(2)
Remarques :
Cette relation est algébrique ;
Si une quantité ( ou ) est effectivement reçue par le système, soit fournie par le milieu extérieur, elle est comptée positivement ;
Si une quantité ( ou ) est cédée par le système au milieu extérieur, elle est comptée négativement.
Puisque cette étude se limite au systèmes fermés, la variation d'énergie cinétique macroscopique est le plus souvent négligeable ou nulle.
Simplification du théorème de l'énergie cinétique
Dans ces conditions, la relation précédente se simplifie :
(3)
C'est sous cette forme que sera dorénavant utilisée la relation exprimant la variation d'énergie interne.
2. Énoncé du premier principe de la thermodynamique
*Remarque préliminaire : il s'agit d'un principe (ou postulat), c'est-à-dire d'une affirmation qui ne se démontre pas mais qui tire sa légitimité de ses conséquences expérimentales.
Ainsi, tant qu'aucun fait expérimental n'est en contradiction avec ce principe, il est considéré comme exact.
Énoncé du premier principe
L'énergie interne d'un système fermé est une fonction d'état.
Cela revient à postuler que l'énergie interne peut simplement s'exprimer en fonction de paramètres d'état simples, comme la masse m, la température T, le volume V et la pression P.
*Conséquence :
La variation d'énergie interne entre un état initial 1 et un état final 2 ne dépend que des paramètres d'état caractérisant l'état 1 et des paramètres d'état caractérisant l'état 2.
Cette variation ne dépend pas de la manière dont s'est opéré la transformation.
Cela est très important du point de vue pratique : imaginons une transformation réelle difficile à modéliser.
Il est possible de contourner la difficulté en calculant la variation réelle d'énergie interne en imaginant un protocole expérimental fictif simple : si ce protocole fictif permet bien de passer de l'état réel 1 à l'état réel 2, la variation d'énergie interne calculée par ce protocole fictif fournit la valeur réelle de la variation d'énergie interne.
*Attention : il résulte de la formule (3) que la valeur de ne dépend pas du « chemin suivi » pour aller de l'état 1 à l'état 2 mais d'une part, d'autre part, dépendent du chemin suivi. Nous reviendrons sur ce point sous forme d'exercice.
Cas limite d'une évolution élémentaire
La variation élémentaire d'énergie interne entre deux instants très proches de dates et est assimilable à une différentielle notée . L'adaptation de la formule (3) à une telle évolution montre que cette variation est égale à une somme de deux quantités élémentaires : la quantité élémentaire de travail, noté , et la quantité élémentaire de chaleur (transfert thermique) notée . D'où l'expression de la variation élémentaire d'énergie interne :
(4)
Remarque : étant une fonction d'état, sa différentielle est une différentielle totale exacte qui vérifie le théorème de Schwarz. Nous reviendrons là-dessus sous forme d'exercice après l'étude du deuxième principe.
TRÈS IMPORTANT : il est fondamental de bien respecter les conventions sur les notations :
Le symbole « » devant le symbole d'une fonction d'état désigne une variation finie ;
Le symbole « » devant le symbole d'une fonction d'état désigne une variation élémentaire, assimilable mathématiquement à une différentielle ;
Le symbole « » devant le symbole d'une quantité désigne une quantité élémentaire.
3. Expression du travail des forces exercées sur le système
3.1. Expression du travail élémentaire
En thermodynamique, le système est presque toujours un fluide, très souvent un gaz.
Le travail reçu par le fluide est alors le travail des forces de pression exercées par les parois sur le fluide.
Remarque préliminaire : si le fluide est enfermé dans une enceinte indéformable, les parois restent fixes, les forces de pression ne travaillent pas : .
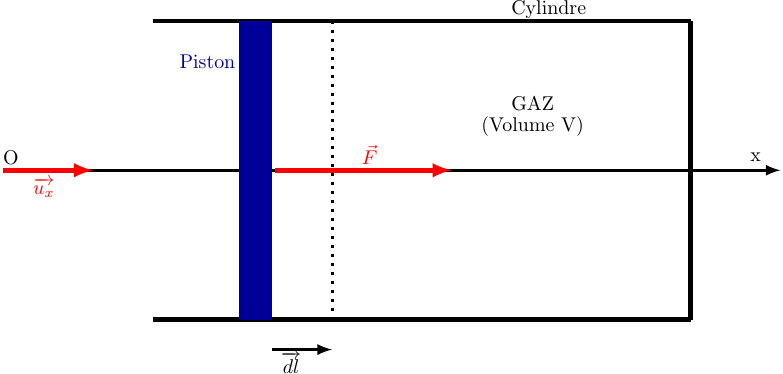
Dans le cas plus général, on imagine un gaz enfermé dans un cylindre dont la section droite est d'aire .
On note l'abscisse de la parois du piston en contact avec le gaz.
* Soit la force exercée par le piston sur le gaz.
* Soit un déplacement élémentaire du piston.
Le travail de la force a pour expression :
(5)
Habituellement, on appelle pression extérieure le rapport (). Ce qui conduit à :
(6)
Dans le cas de la figure, on peut remarquer que le produit représente la diminution de volume du gaz, soit l'opposé de sa variation dV. D'où l'expression classique du travail élémentaire :
(7)
Cette formule est algébrique et valide quel que soit le sens de déplacement du piston : imaginons que le pistons se déplace vers la gauche.
Le travail serait alors négatif puisque vecteur force et vecteur déplacement seraient de sens opposés.
Cela correspondrait à une augmentation de volume : donc : .
Attention : la pression extérieure est le rapport () ; il ne s'agit pas nécessairement de la pression ambiante existant à l'extérieur du cylindre ;
cette dernière étant en général la pression atmosphérique .
Dans toutes la suite de ce document, nous supposerons que seules les forces de pression sont susceptibles de travailler.
3.2. Expression du travail
Dans le cas général, il faut intégrer l'expression précédente :
(8)
L'expression n'est pas nécessairement simple si la force exercée par le piston varie au court de son déplacement.
3.3. Cas particulier: Pext = constante
L'expression du travail devient simple :
(9)
Ce cas particulier est, en pratique, extrêmement fréquent :
il concerne les gaz enfermés dans un cylindre avec un piston mobile sans frottement de masse négligeable.
Alors : et la pression atmosphérique peut être considérée comme fixe pendant la durée de la transformation.
Il concerne aussi les liquides et les solides laissés à l'air libre ou enfermés dans des récipients non étanches.
En absence d'évaporation, le système peut être considéré comme fermé et là encore : .
Cette situation concerne par exemples les évolutions dans les calorimètres usuels et un grand nombre de transformations chimiques.
3.4. Cas particulier d'une transformation quasi statique
Cette approximation est très souvent utilisée en mécanique, lorsqu'il s'agit d'étudier des systèmes complexes évoluant à vitesses et accélérations faibles.
On considère les produits () suffisamment faibles pour qu'à chaque instant, on puisse appliquer les lois de la statique, plus simples que les lois de la dynamique.
De même en thermodynamique, une transformation peut être qualifiée de quasi statique s'il est possible, en bonne approximation, de la décrire comme une suite continue d'états extrêmement proches d'états d'équilibre.
Chaque état doit être considéré comme un état d'équilibre interne.
Cela signifie que, à un instant donné, le système peut être caractérisé par un volume donné, une pression donnée (la même en tout point du système) et une température donnée (la même en tout point du système), ces paramètres d'état étant reliées entre eux par une équation d'état ( pour un gaz parfait par exemple).
Suivant le contexte expérimental, le système doit aussi être, en bonne approximation, en équilibre avec le milieu extérieur.
Prenons deux exemples :
*Exemple 1 : gaz à la température initiale enfermé dans un récipient aux parois diathermanes (perméable à la chaleur) entouré d'un milieux extérieur à la température .
Pour qu'un échange thermique soit possible entre le système et le milieu extérieur, un écart de température doit exister mais celui-ci doit être extrêmement petit de façon que le transfert thermique soit très lent.
Sinon, au cours de l'échange, le gaz au voisinage des parois n'est pas à la même température que le gaz éloigné des parois ; il n'est pas possible de considérer le gaz homogène à une température et une pression données.
Le transfert thermique quasi statique doit donc être très lent avec .
Remarque : en cas de système thermiquement isolé de l'extérieur par des parois athermanes, la transformation est adiabatique (). La contrainte disparaît.
L'évolution doit cependant toujours s'effectuer très lentement pour que la température puisse être considérée comme la même à un instant donné en tout point du système.
*Exemple 2 : On reprend comme système le gaz enfermé dans le cylindre du §3.1 mais le cylindre est maintenant vertical, l'axe étant orienté vers le bas.
Comment réaliser une compression quasi statique à partir d'un état initial de pression ? Supposons que l'on pose sur le piston une surcharge importante de façon à augmenter brutalement ?
Le principe des actions réciproques (principe de l'action et de la réaction comme on dit encore) affirme : .
Cela impose une pression du gaz égale à au contact avec le piston mais cela ne dit rien sur la pression en tout point du gaz à l'intérieur du cylindre.
L'étude rigoureuse est complexe et fait intervenir la propagation des ondes dans les gaz.
On peut ici se contenter d'affirmer que la compression est beaucoup trop rapide pour que la pression du gaz soit, à un instant donné, la même en tout point du gaz.
La compression rapide n'est donc pas quasi statique.
On peut rendre la transformation quasi statique en rajoutant la surcharge très progressivement sous forme de petites masselottes ajoutées les unes après les autres en laissant le temps au gaz, entre chaque ajout, de devenir homogène et au piston de s'immobiliser.
On pourra dans ces conditions affirmer que, au cours de l'évolution :P.
Là encore, la transformation quasi statique doit être très lente.
Important
Lors d'une transformation quasi statique, le travail des forces de pression peut s'écrire :
(10)
4. Une autre fonction d'état : l'enthalpie H
4.1. Définition
Remarque préliminaire : étant une fonction d'état, on peut définir une autre fonction d'état en ajoutant ou retranchant à une expression ne faisant intervenir que des paramètres d'état sous réserve que cette expression est la même dimension que : celle d'une énergie.
C'est le cas du produit .
Définition
On définit la fonction d'état enthalpie du système fermé de la façon suivante :
(11)
4.2. Applications aux transformations monobares
Propriété
Une transformation monobare à les deux propriétés suivantes :
1° : La pression reste fixe au cours de la transformation ;
2° : l'état initial et l'état final sont deux états d'équilibre mécanique.
Pour résumer :
avec fixe au cours de la transformation.
Dans ces conditions :
En tenant compte des relations (3) et (9) :
Donc, pour une transformation monobare, quasi statique ou non :
(12)
Cette relation est particulièrement importante puisque les transformations monobares sont très fréquentes en pratique.
Il suffit de reprendre les conditions du paragraphe 3.3 avec comme état initial et comme état final deux états d'équilibre mécaniques.
Pour ces transformations, il est possible de calculer comme une variation de fonction d'état. Lorsque la transformation est délicate à modéliser, il est possible d'imaginer une succession d'étapes fictives simples qui vont de l'état initial réel à l'état final réel.
C'est, par exemple, ce que l'on fait pour étudier la fonte d'un glaçon dans un calorimètre sous pression atmosphérique fixe.
On calcule la quantité de chaleur Q comme une somme de quantités de chaleurs correspondant à une succession de transformations simples :
échauffement du glaçon jusqu'à 0°C ; fusion du glaçon à température fixe : 0°C ; échauffement de la « glace fondue » jusqu'à la température finale.
Ce raisonnement serait faux si Q ne correspondait pas à une variation de fonction d'état.
Remarque 1 : le résultat (12) est aussi valide pour une transformation isobare quasi statique. Pour le montrer, je détermine la différentielle de définie par (11) :
L'évolution étant quasi statique, la relation (10) conduit à :
(13)
Cas particulier de l'évolution isobare :
(14)
Remarque 2 : il existe encore une autre situation où un transfert thermique peut s'évaluer comme une variation de fonction d'état ; il s'agit de la transformation isochore. De façon évidente, la relation (7) conduit à :
(15)
5. Capacités thermiques
5.1. Généralités
Les mesures de calorimétrie, réalisées sur de faibles intervalles de températures, montrent que le transfert thermique reçu par un corps pur est proportionnel à la variation de température T, à la quantité de matière du système.
Il dépend aussi de la nature du corps pur.
On obtient ainsi, pour une quantité « » de matière, une expression de la forme générale :
(16)
avec : capacité thermique (ou calorifique) molaire, propriété caractéristique du corps pur concerné.
Il est aussi possible de définir une capacité thermique massique « » telle que :
(17)
Il en résulte : , soit : où « » désigne la masse molaire du corps pur.
On définit également la capacité thermique du système :
(18)
Cette première approche mérite d'être affinée pour deux raisons :
* L'étude sur des intervalles de température importants montre qu'il faut en réalité considérer la capacité thermique comme une fonction de la température ;
* Le transfert thermique dépend de la manière dont il est réalisé : il faut distinguer les transferts isochores des transfert isobares. Il faut donc une définition plus rigoureuses des capacités thermiques.
5.2. Définitions des capacités thermiques
5.2.1 - Capacité thermique isobare Cp
Compte tenu de la relation (15), pour un transfert thermique isobare élémentaire :
(19)
D'où une expression générale de la capacité thermique isobare :
(20)
5.2.2 - Capacité thermique isochore Cv
Compte tenu de la relation (16), pour un transfert thermique isochore élémentaire :
(21)
D'où une expression générale de la capacité thermique isochore :
(22)
5.2.3 - Cas particulier des gaz parfaits
Un gaz parfait : cela n'existe pas dans la réalité.
Il s'agit juste d'un modèle simplifié de la réalité qui tient compte de l'énergie cinétique d'agitation thermique mais néglige l'influence des forces d'interactions entre les molécules.
Cependant, ce modèle constitue une approximation d'autant meilleure que la pression est faible et les conditions de température et de pression éloignées des conditions de liquéfaction du gaz.
Pour les gaz monoatomiques et les gaz diatomiques contenus dans l'air, l'approximation du gaz parfait est excellente aux températures pas trop faibles (supérieures à 150K environ) et pour des pressions ne dépassant pas dix fois la pression atmosphérique (pressions inférieures à 1 MPa environ).
Du point de vue macroscopique, un gaz parfait possède deux propriétés :
1° : À l'équilibre, il vérifie l'équation d'état : ;
2° : il vérifie la première loi de Joule :
Première loi de Joule
Son énergie interne ne dépend que de T.
Remarque : si un gaz parfait vérifie la première loi de Joule, il vérifie aussi la seconde loi de Joule :
Deuxième loi de Joule
Son son enthalpie ne dépend que de T.
En effet, pour un gaz parfait :
(23)
Les lois de Joule permettent de fournir des définitions simplifiées des capacités thermiques pour les gaz parfaits :
En tenant compte de (23) :
(24)
Ce résultat est connu sous le nom de relation de Mayer.
Cette relation peut s'appliquer aux capacités thermiques molaires en divisant tous les termes par :
(25)
Il est fréquent de faire intervenir le coefficient de Laplace :
(26)
De la relation de Mayer, on peut déduire les expressions des capacités thermiques en fonction de R et du coefficient de Laplace. La relation (25) devient :
D'où les expressions des capacités thermiques molaires d'un gaz parfait :
(27)
La thermodynamique statistique fournit des valeurs du coefficient de Laplace pour les gaz parfaits monoatomiques (gaz rares) et les gaz parfaits diatomiques :
(28)
On en déduit des expressions simples de et de pour ces gaz qui bien sûr vérifient les lois de Joule :
Gaz parfait monoatomique :
Gaz parfait diatomique :
Les constantes peuvent être choisies arbitrairement puisque seules les variations d'énergie interne et d'enthalpie ont un sens physique.
5.2.4 - Cas des solides et des liquides
Les propriétés thermodynamiques des liquides et des solides dépendent très peu de la pression sauf dans des cas non étudiés ici où les variations de pressions sont très importantes.
Dans le cadre de cette étude, nous négligerons cette influence, ce qui nous conduit à ne pas faire la différence entre capacité thermique isobare et capacité thermique isochore.
Nous noterons simplement la capacité thermique d'un solide ou d'un liquide.
Cela revient à conserver les notations de l'étude préliminaire du §5.1.
6. Transformation quasi statique et adiabatique d'un gaz parfait : lois de Laplace
Il est important de noter d'emblée les quatre conditions nécessaires à la validité de cette loi :
1° : seules les forces de pression sont susceptibles de travailler ;
2° : évolution quasi statique ;
3° : gaz assimilé à un gaz parfait ;
4° : évolution adiabatique : aucun transfert thermique n'intervient lors de l'évolution, ce qui suppose les parois du cylindre et du piston athermanes, c'est à dire suffisamment bien isolées thermiquement du milieu extérieur.
Bien sûr : ces conditions ne peuvent dans la réalité être obtenues que de façon très approchée.
Première loi de Joule en tenant compte de (28) :
Transformation adiabatique :
Ce qui conduit à :
En intégrant, on obtient :
Soit encore :
(29)
La transformation étant quasi statique, l'équation d'état des gaz parfaits est vérifiée pour chaque état intermédiaire.
Il est donc possible de remplacer dans (29) T par son expression déduite de l'équation d'état :
;
d'où une autre expression de la loi de Laplace, équivalente mais plus simple à retenir :
(30)
Il est aussi possible de remplacer dans (30) V par son expression déduite de l'équation d'état :
;
d'où une troisième expression de la loi de Laplace.
Bien sûr, dans les trois expressions, les constantes sont différentes !
(31)
7. Exercice d'illustration
7.1. Énoncé
On dispose de d'un gaz parfait diatomique () dans un état initial 1 caractérisé par un volume sous la pression .
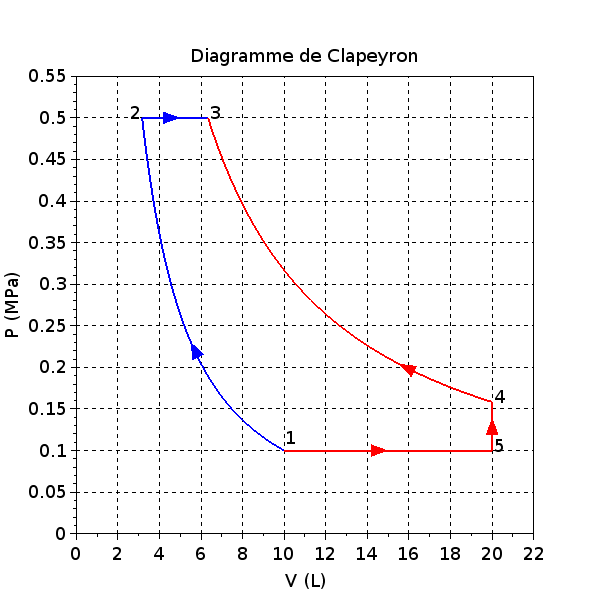
On se propose de l'amener à un état final 3 par deux « chemins » différents représentés ci-dessus dans le diagramme de Clapeyron P = f(V) :
Premier chemin en deux étapes (en bleu dans le diagramme) :
- passage de l'état 1 à un état intermédiaire 2 par une compression adiabatique avec .
- passage de l'état 2 à l'état final 3 par un échauffement isobare qui conduit à un volume V3 = 2V2.
Second chemin en trois étapes (en rouge dans le diagramme) :
- passage de l'état 1 à un état 5 par un échauffement isobare jusqu'à un volume V5 = 2V1.
- passage de l'état 5 à un état 4 par une compression isochore jusqu'à une pression P4.
- passage de l'état 4 à l'état final 3 par une compression isotherme.
1° : Déterminer, pour chacun des cinq états, les paramètres d'état manquants : P, T, V.
2° : Pour chacune des transformations, déterminer le transfert thermique reçu par le gaz (Q) et le travail reçu (W).
3° : En déduire QB et WB : transfert thermique et travail pour aller de l'état initial 1 à l'état final 3 par le premier chemin.
4° : En déduire QR et WR : transfert thermique et travail pour aller de l'état initial 1 à l'état final 3 par le second chemin.
5° : Expliquer en quoi ces résultats illustrent le contenu de cette fiche.
7.2. Corrigé
1° : L'état 1 vérifie l'équation d'état des gaz parfaits :
Application numérique :
La loi de Laplace s'applique à l'évolution 1-2 :
Application numérique :
Cet état 2 vérifie aussi l'équation d'état des gaz parfaits :
L'état 3 vérifie aussi l'équation d'état des gaz parfaits avec : et
;
Application numérique :
Concernant l'état 4 : on sait : et on sait que l'évolution 4-3 est isotherme.
Cela conduit à :
et :
soit :
Application numérique :
Tous ces paramètres d'états peuvent être rassemblés dans un tableau :
États
1
2
3
4
5
P (MPa)
0,100
0,500
0,500
0,158
0,100
V (L)
10
3,17
6,34
20
20
T (K)
241
381
762
762
481
2° La transformation 1-2 est adiabatique : Q12 =0
Pour W12, il est possible d'intégrer l'expression du travail (10) en tenant compte de la loi de Laplace.
Il est beaucoup plus rapide d'utiliser la relation (3) couplée avec la première loi de Joule.
Application numérique :
La transformation 2-3 est isobare ; le travail se calcule aisément :
Application numérique :
Concernant Q23, le plus rapide consiste à utiliser la relation (14) et la seconde loi de Joule :
Application numérique :
La transformation 1-5 est isobare comme la transformation 2-3. Le raisonnement est le même.
La transformation 5-4 est isochore. De façon immédiate : W54 = 0
Il en résulte :
Application numérique :
La transformation 4-3 est isotherme. Au cours de la transformation quasi statique, pour chaque état intermédiaire : . La relation (10) devient :
Application numérique :
Selon la première loi de Joule, la variation d'énergie interne est nulle lors d'une transformation isotherme.
3° Concernant le chemin « bleu » sur le diagramme pour aller de l'état 1 à l'état 3, nous avons :
4° Concernant le chemin « rouge » sur le diagramme pour aller de l'état 1 à l'état 3 :
On remarque : et : un transfert thermique et une quantité de travail ne se calculent pas, dans le cas général, comme des variations de fonctions d'état.
Leurs valeurs, entre deux états donnés dépendent de la manière dont s'est effectuée la transformation.
En revanche, la somme de ces deux quantités est indépendante du « chemin suivi » car égale à la variation de l'énergie interne qui est une fonction d'état.
On le vérifie aisément en remarquant :
Remarque : tous les résultats sont affichés avec la même précision que les données : trois chiffres significatifs. Cependant, pour éviter que des arrondis successifs ne finissent par induire une erreur significative, les résultats intermédiaires sont conservés en mémoire avec le maximum de précision autorisée par la calculatrice.
Publié par gbm / relue gbm
le
ceci n'est qu'un extrait
Pour visualiser la totalité des cours vous devez vous inscrire / connecter (GRATUIT) Inscription Gratuitese connecter
Merci à vanoise pour avoir contribué à l'élaboration de cette fiche
Désolé, votre version d'Internet Explorer est plus que périmée ! Merci de le mettre à jour ou de télécharger Firefox ou Google Chrome pour utiliser le site. Votre ordinateur vous remerciera !
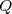 .
.
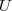 , du système.
Cette énergie interne est une somme de deux termes :
, du système.
Cette énergie interne est une somme de deux termes :
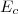 qui s'intéresse au mouvement d'ensemble des particules par rapport au repère d'étude.
qui s'intéresse au mouvement d'ensemble des particules par rapport au repère d'étude.
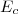 due à l'écoulement et une énergie cinétique d'agitation thermique prise en compte dans la valeur de l'énergie interne
due à l'écoulement et une énergie cinétique d'agitation thermique prise en compte dans la valeur de l'énergie interne 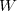 ou
ou  Cela revient à postuler que l'énergie interne peut simplement s'exprimer en fonction de paramètres d'état simples, comme la masse m, la température T, le volume V et la pression P.
Cela revient à postuler que l'énergie interne peut simplement s'exprimer en fonction de paramètres d'état simples, comme la masse m, la température T, le volume V et la pression P.
) ne dépend pas du « chemin suivi » pour aller de l'état 1 à l'état 2 mais
ne dépend pas du « chemin suivi » pour aller de l'état 1 à l'état 2 mais 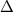 » devant le symbole d'une fonction d'état désigne une variation finie ;
» devant le symbole d'une fonction d'état désigne une variation finie ;
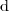 » devant le symbole d'une fonction d'état désigne une variation élémentaire, assimilable mathématiquement à une différentielle ;
» devant le symbole d'une fonction d'état désigne une variation élémentaire, assimilable mathématiquement à une différentielle ;
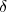 » devant le symbole d'une quantité désigne une quantité élémentaire.
» devant le symbole d'une quantité désigne une quantité élémentaire.
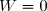 .
.
 .
On note
.
On note  l'abscisse de la parois du piston en contact avec le gaz.
l'abscisse de la parois du piston en contact avec le gaz.
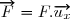 la force exercée par le piston sur le gaz.
la force exercée par le piston sur le gaz.
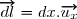 un déplacement élémentaire du piston.
Le travail de la force a pour expression :
un déplacement élémentaire du piston.
Le travail de la force a pour expression :
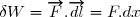 (5)
(5)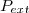 le rapport (
le rapport (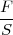 ). Ce qui conduit à :
). Ce qui conduit à :
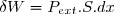 (6)
(6)) représente la diminution de volume du gaz, soit l'opposé de sa variation dV. D'où l'expression classique du travail élémentaire :
représente la diminution de volume du gaz, soit l'opposé de sa variation dV. D'où l'expression classique du travail élémentaire :
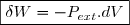 (7)
(7)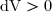 donc :
donc : 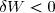 .
.
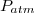 .
.
 (8)
(8) = - P_{ext} . \Delta V}) (9)
(9)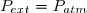 et la pression atmosphérique peut être considérée comme fixe pendant la durée de la transformation.
et la pression atmosphérique peut être considérée comme fixe pendant la durée de la transformation.
 ) suffisamment faibles pour qu'à chaque instant, on puisse appliquer les lois de la statique, plus simples que les lois de la dynamique.
De même en thermodynamique, une transformation peut être qualifiée de quasi statique s'il est possible, en bonne approximation, de la décrire comme une suite continue d'états extrêmement proches d'états d'équilibre.
) suffisamment faibles pour qu'à chaque instant, on puisse appliquer les lois de la statique, plus simples que les lois de la dynamique.
De même en thermodynamique, une transformation peut être qualifiée de quasi statique s'il est possible, en bonne approximation, de la décrire comme une suite continue d'états extrêmement proches d'états d'équilibre.
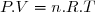 pour un gaz parfait par exemple).
pour un gaz parfait par exemple).
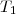 enfermé dans un récipient aux parois diathermanes (perméable à la chaleur) entouré d'un milieux extérieur à la température
enfermé dans un récipient aux parois diathermanes (perméable à la chaleur) entouré d'un milieux extérieur à la température 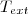 .
Pour qu'un échange thermique soit possible entre le système et le milieu extérieur, un écart de température doit exister mais celui-ci doit être extrêmement petit de façon que le transfert thermique soit très lent.
Sinon, au cours de l'échange, le gaz au voisinage des parois n'est pas à la même température que le gaz éloigné des parois ; il n'est pas possible de considérer le gaz homogène à une température
.
Pour qu'un échange thermique soit possible entre le système et le milieu extérieur, un écart de température doit exister mais celui-ci doit être extrêmement petit de façon que le transfert thermique soit très lent.
Sinon, au cours de l'échange, le gaz au voisinage des parois n'est pas à la même température que le gaz éloigné des parois ; il n'est pas possible de considérer le gaz homogène à une température 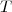 et une pression
et une pression 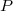 données.
Le transfert thermique quasi statique doit donc être très lent avec
données.
Le transfert thermique quasi statique doit donc être très lent avec 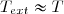 .
.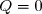 ). La contrainte
). La contrainte ) étant orienté vers le bas.
Comment réaliser une compression quasi statique à partir d'un état initial de pression
étant orienté vers le bas.
Comment réaliser une compression quasi statique à partir d'un état initial de pression  ? Supposons que l'on pose sur le piston une surcharge importante de façon à augmenter brutalement
? Supposons que l'on pose sur le piston une surcharge importante de façon à augmenter brutalement 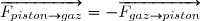 .
.
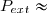 P.
Là encore, la transformation quasi statique doit être très lente.
P.
Là encore, la transformation quasi statique doit être très lente.
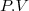 .
.

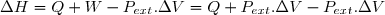
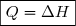 (12)
(12)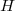 définie par (11) :
définie par (11) :
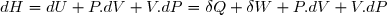
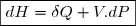 (13)
(13) (14)
(14)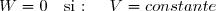
 (15)
(15) T, à la quantité de matière du système.
Il dépend aussi de la nature du corps pur.
On obtient ainsi, pour une quantité «
T, à la quantité de matière du système.
Il dépend aussi de la nature du corps pur.
On obtient ainsi, pour une quantité « 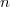 » de matière, une expression de la forme générale :
» de matière, une expression de la forme générale :
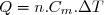 (16)
(16)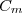 : capacité thermique (ou calorifique) molaire, propriété caractéristique du corps pur concerné.
Il est aussi possible de définir une capacité thermique massique «
: capacité thermique (ou calorifique) molaire, propriété caractéristique du corps pur concerné.
Il est aussi possible de définir une capacité thermique massique «  » telle que :
» telle que :
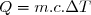 (17)
(17)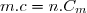 , soit :
, soit : 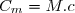 où «
où « 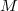 » désigne la masse molaire du corps pur.
» désigne la masse molaire du corps pur.
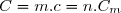 (18)
(18)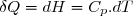 (19)
(19)_{P}}) (20)
(20)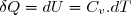 (21)
(21)_{V}}) (22)
(22)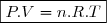 ;
;
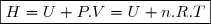 (23)
(23)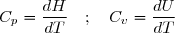
}{dT} = C_{v} + n . R) (24)
(24) (25)
(25)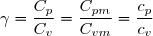 (26)
(26)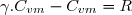
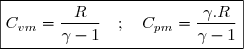 (27)
(27)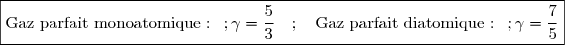 (28)
(28)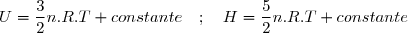
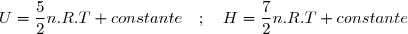
 la capacité thermique d'un solide ou d'un liquide.
Cela revient à conserver les notations de l'étude préliminaire du §5.1.
la capacité thermique d'un solide ou d'un liquide.
Cela revient à conserver les notations de l'étude préliminaire du §5.1.
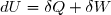
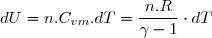
 \cdot \dfrac{dV}{V} = 0}})
 + \left(\gamma -1 \right) \cdot \ln \left(V \right) = constante)
} = constante}) (29)
(29)} = \dfrac{P . V^{\gamma}}{n . R} = constante) ;
d'où une autre expression de la loi de Laplace, équivalente mais plus simple à retenir :
;
d'où une autre expression de la loi de Laplace, équivalente mais plus simple à retenir :
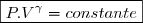 (30)
(30)^{\gamma} = \left(n . R \right)^{\gamma} \cdot \dfrac{T^{\gamma}}{P^{\left(\gamma - 1 \right)}} = constante) ;
d'où une troisième expression de la loi de Laplace.
Bien sûr, dans les trois expressions, les constantes sont différentes !
;
d'où une troisième expression de la loi de Laplace.
Bien sûr, dans les trois expressions, les constantes sont différentes !
}}) (31)
(31)
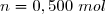 d'un gaz parfait diatomique (
d'un gaz parfait diatomique (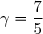 ) dans un état initial 1 caractérisé par un volume
) dans un état initial 1 caractérisé par un volume 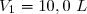 sous la pression
sous la pression 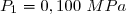 .
On se propose de l'amener à un état final 3 par deux « chemins » différents représentés ci-dessus dans le diagramme de Clapeyron P = f(V) :
.
On se propose de l'amener à un état final 3 par deux « chemins » différents représentés ci-dessus dans le diagramme de Clapeyron P = f(V) :
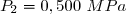 .
.
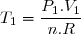
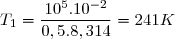
^{\dfrac{1}{\gamma}})
^{\dfrac{5}{7}}=3,17 ~L)
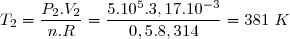
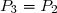 et
et 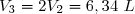
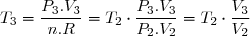 ;
;
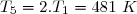
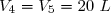 et on sait que l'évolution 4-3 est isotherme.
Cela conduit à :
et on sait que l'évolution 4-3 est isotherme.
Cela conduit à :
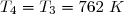 et :
et : 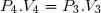 soit :
soit : 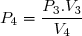
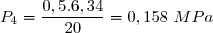
= \dfrac{5}{2} . n . R . \left(T_{2} - T_{1} \right))
 = 1,46. 10^{3} ~ J)
)
 = -1,58.10^{3} ~ J)
)
 = 5,54.10^{3} ~ J)
 \quad ; \quad W_{15} = -10^{5} . \left(2.10^{-2}-10^{-2}\right) = -1,00.10^{3} ~J)
\quad ; \quad Q_{15}=3,5.0,5.8,314.\left(481-241\right) = 3,50.10^{3} ~ J)
 = \dfrac{5}{2}.n.R. \left(T_{4}-T_{5} \right))
 = 2,92.10^{3} ~J)
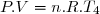 . La relation (10) devient :
. La relation (10) devient :)
 = 3,64.10^{3} ~ J)
 - W_{43} = - W_{43} \quad ; \quad Q_{43}= -3,64.10^{3} ~ J)
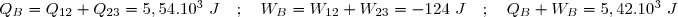
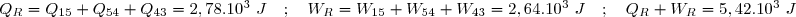
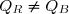 et
et 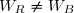 : un transfert thermique et une quantité de travail ne se calculent pas, dans le cas général, comme des variations de fonctions d'état.
Leurs valeurs, entre deux états donnés dépendent de la manière dont s'est effectuée la transformation.
En revanche, la somme de ces deux quantités est indépendante du « chemin suivi » car égale à la variation de l'énergie interne qui est une fonction d'état.
On le vérifie aisément en remarquant :
: un transfert thermique et une quantité de travail ne se calculent pas, dans le cas général, comme des variations de fonctions d'état.
Leurs valeurs, entre deux états donnés dépendent de la manière dont s'est effectuée la transformation.
En revanche, la somme de ces deux quantités est indépendante du « chemin suivi » car égale à la variation de l'énergie interne qui est une fonction d'état.
On le vérifie aisément en remarquant :)
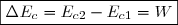 (1)
(1)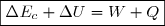 (2)
(2)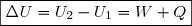 (3)
(3)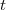 et
et ) est assimilable à une différentielle notée
est assimilable à une différentielle notée 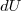 . L'adaptation de la formule (3) à une telle évolution montre que cette variation est égale à une somme de deux quantités élémentaires : la quantité élémentaire de travail, noté
. L'adaptation de la formule (3) à une telle évolution montre que cette variation est égale à une somme de deux quantités élémentaires : la quantité élémentaire de travail, noté 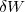 , et la quantité élémentaire de chaleur (transfert thermique) notée
, et la quantité élémentaire de chaleur (transfert thermique) notée 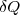 . D'où l'expression de la variation élémentaire d'énergie interne :
. D'où l'expression de la variation élémentaire d'énergie interne :
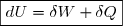 (4)
(4)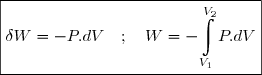 (10)
(10)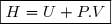 (11)
(11)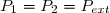 avec
avec  forums de physique - chimie
forums de physique - chimie