Inscription / Connexion Nouveau Sujet
thermodynamique
Bonjour, j'essaye de faire cette exercice mais je bloque sur certains points.
Partie 1
1. Bouilloire électrique
Une bouilloire électrique, de puissance 1000W, est utilisée pour porter à
ébullition 600 mL d'eau initialement à 20°C et à la pression de 1 bar. La
bouilloire est parfaitement isolée thermiquement et l'énergie électrique est
intégralement transmise à l'eau.
1.1. Quelle quantité d'eau liquide reste-t-il dans la bouilloire après 5
minutes de chauffage ?
1.2. Calculer la variation d'entropie des 600 mL d'eau pendant cette
durée.
Données : Cp,m (H20,l)=75.2 J/K/mol
Chaleur latente de vaporisation L l->v (373K)=40.6 kJ/mol
2. Entropie molaire du méthanol liquide
On cherche à connaitre l'entropie molaire absolue du méthanol gazeux sous une
pression de 1 bar à 298K. Cette donnée ne peut être directement mesurée car le
méthanol gazeux n'existe pas sous une pression de 1 bar. Elle doit donc être
calculée. On admettra que le méthanol liquide est incompressible et que le
méthanol gazeux est un gaz parfait.
2.1. Par quelle suite d'étapes réversibles peut-on passer du méthanol
liquide à 298 K et 1 bar au méthanol gazeux aux mêmes pression et
température sachant que la pression de vapeur saturante du méthanol à
298 K est de 0.1651 bar?
2.2. A l'aide du cycle précédent et des données, en déduire l'entropie
molaire absolue du méthanol gazeux sous une pression de 1 bar à 298K.
Données : Chaleur latente de vaporisation L l->v (298K)=37.3 kJ/mol, Entropie
molaire absolue du méthanol liquide
0
meth liq ,
S =127.8 J/mol/
Partie2
1. Grandeur de mélange
Une grandeur (extensive) X de mélange, notée ΔmélX, est définie comme la
différence ΔmélX=Xf-X0 où :
- Xf est la grandeur du système monophasé du mélange à la température T et
sous la pression P,
la grandeur molaire du constituant Bi pur à la
température T et sous la pression P et ni sa quantité de matière.
1.1. Mélange toluène/benzène.
Prenons un mélange entre deux hydrocarbures. A 298 K, 234 g de benzène et
184g de toluène sont mélangés. Le mélange formé est idéal. La quantité de ces
hydrocarbures sous forme vapeur, en équilibre avec le liquide, sera négligé
devant celle de ces hydrocarbures dans le liquide.
1.1.1.Calculer la fraction molaire de chacun des constituants
1.1.2.Soit dans un premier temps X=G. Exprimer G0 puis Gf.
1.1.3. En déduire l'expression, puis la valeur de ΔmélG. Commenter.
1.1.4.A partir de la différentielle exacte de G, en déduire ΔmélV et ΔmélS.
1.1.5.A partir de la relation de Gibbs-Helmoltz, calculer ΔmélH.
1.1.6.Commenter ces valeurs.
Données : M (benzène)= 78 g/mol ; M(toluène)= 92 g/mol
1.2. Grandeurs molaires de mélange
1.2.1. Donner, pour un mélange idéal, l'expression générale de l'enthalpie
libre molaire de mélange, de deux constituants B1 et B2 en fonction de
la fraction molaire x2 du constituant B2.
1.2.2.Chercher pour quelle fraction molaire x2, ΔmélGm est extrémale.
Commenter.
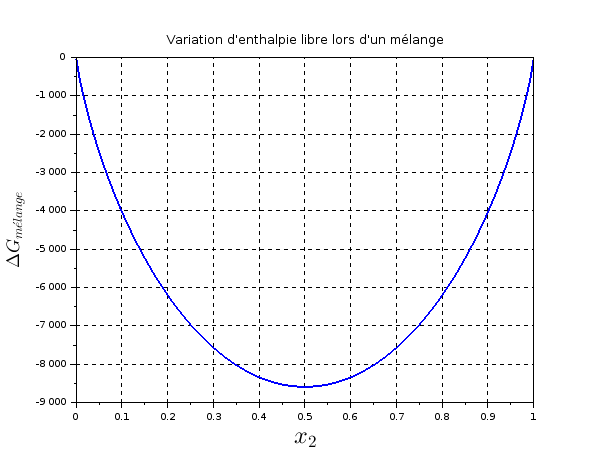
1.2.3.Représenter les variations de ΔmélGm en fonction de x2.
2. Grandeur d'excès
L'écart à l'idéalité d'une solution est estimé par mesure des grandeurs d'excès,
définies par la différence entre les grandeurs caractéristiques du mélange réel et
les grandeurs du mélange idéal correspondant (ici, la solution idélae). Elles sont
notées avec l'exposant E. Les coefficients d'activité
, relatifs aux
fractions molaires, sont définis à partir de l'expression du potentiel chimique
par :
2.1. Exprimer, pour un mélange réel, l'enthalpie libre molaire de
mélange ΔmélGm.
2.2. Montrer que l'enthalpie libre molaire d'excès est de la forme
Pour la partie 1:
1.1)J'ai trouve qu'il restait 556mL dans la bouillir, j'ai multiplie la puissance par 5 minute ( 300 000 J) j'ai calcule l'enthalpie pour chauffe l'eau.
j'ai Ht=H(chauffe)+nL
j'isole n et je trouve la quantite 'eau
1.2) S=871,74 J
2) je bloque ici
Bonjour
OK pour 1.1
1.2 : OK pour le calcul mais il s'agit d'une variation  S et il ne faut pas oublier l'unité.
S et il ne faut pas oublier l'unité.
2.L'entropie étant une fonction d'état ; on peut imaginer un chemin réversible pour passer de l'état initial à l'état final mais il ne s'agit pas d'une évolution cyclique comme indiqué dans l'énoncé à la question suivante. Tu peux donc imaginer la succession d'évolutions réversibles suivante :
méthanol liquide à 298K sous 1bar  méthanol liquide sous 0,1651bar à 298K méthanol gaz sous 0,1651bar à 298Kméthanol gazeux sous 1bar à 298K
méthanol liquide sous 0,1651bar à 298K méthanol gaz sous 0,1651bar à 298Kméthanol gazeux sous 1bar à 298K
J'ai posé S ( la variation d'entropie du changement d'etat l-->g) et S1= entropie l(1bar)-->l(0,1651 bar)
S2= entropie l(0,1651 bar)---> g(0,1651 bar)
S3= entropie g(0,1651bar)--->g(1bar)
S=S1+S2+S3
S2= L/T= 125,17 J/K/mol
j'ai du mal pour le reste
Pour une évolution élémentaire réversible quelconque d'un corps pur de masse fixe, on obtient :
Le liquide est supposé incompressible : les variations de V en fonction de T sont supposées nulles ; h=0. On peut donc considérer que la compression ou la détente réversible isotherme du liquide se fait sans échange de chaleur. Donc S a l'expression la plus simple imaginable...
la reaction est isotherme donc Q= Cp T=0 , si je pose S=Q/T alors S=0 pour les deux reaction de compression et de dilatation?
Ton message de 15h39 est parfait.
Ton message de 16h conduit au même résultat puisque, à T fixe, V est fixe :
W=- P.dV=0 donc S=0.
P.dV=0 donc S=0.
Cependant, il faut supposer que U ne dépend que de T. Cela est exact pour un gaz parfait mais l'énoncé ne précise pas que l'approximation est valide pour un liquide pur indilatable, même si cela est vrai.
pour conclure on pose que Sm(g) = 125, 17 J/L/mol ? je trouve que c'est peu je m'attendais a quelque chose de beaucoup plus grand qu' en phase liquide
Tu as commis deux erreurs :
1° : si la variation d'entropie du liquide par variation isotherme de pression est nulle, il n'en est pas de même pour la vapeur que l'énoncé demande d'assimiler à un gaz parfait.
2° : on demande l'entropie absolue : il faut donc ajouter à la variation d'entropie celle du liquide dont la valeur est fournie par l'énoncé.
(1) phase gaz à 0,1651 bar et (2) phase gaz à 1bar
je pose S=-W/T or on sait que P(1)V(1)=nRT et P(2)V(2)=nRT --> P(1)V(1)=P(2)V(2)
Je pose que le volume molaire d'un gaz parfait est de 24L/mol (pour une mol)
j'obtient un travail de 4322,89 J/K/mol
S(w)=-W/T=-14,51 J/K/mol
je pose St= S*(liquide)+S(w)+S(gaz) et je n'arrive pas a conclure
On sait que St= S(gaz)- S(liq)=L/T+S(w)
donc S(g)= S(liq)+L/T+S(w)= 127,8 +125,17-14,51 = 238,46 J/K/mol
pour la partie2:
1)je pose x1 pour la fraction molaire du benzene et x2 pour celle du toluene
x1=3/5 et x2=2/5
2) G0=n1*G1,m+n2*G2,m = n1*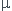 1+n2*2
1+n2*2
mais je n'arrive pas a definir Gf, j'ai du mal avec la definition donne dans l'enonce
Le début est bien vu mais la suite est maladroite puisque l'objectif est de calculer, pour un compression isotherme réversible d'un gaz parfait :
Comme tu l'as écris :
Donc :
Ici : n=1mol. Ne confond pas entropie S et variation d'entropie S...
Bonjour je trouve S3= RTln(P2/P1)= - 14,98 J/K/mol
Je somme toutes les entropie
S(g)=S(l)+S2+S3 = 237,99 J/K/mol
La valeur est pas loin du double de celle du liquide, le resultat est coherent.
Pour la partie 2: (1) benzene et (2) toluene
1.1.1 n(1)=3 mol et n(2)=2 mol
donc x(1)=3/5 et x2=2/5
1.1.2
= -8337,17J <0
la variation d'enthalpie libre est négatif donc cette reaction est spontanée ( ces deux espece se melangent bien?)
1.1.4
Pour le volume j'ai du mal mais pour l'entropie je derive par rapport a T l'equation de G et j'obtient S = -n(1)Rln(x(1))-n(2) Rln(x(2))= 27,977 J/K
1.1.5
OK jusqu'à 1.1.2
Pour la suite, le document suivant, en particulier pages 10 et suivantes devrait t'aider (leçon 3) :
![]()
Je n'arrive toujours pas a deduire V, (pour moi il sera trés petit et donc negligeable )
1.1.4
HG-TS
Or TS=- G donc H=0
1.1.5
G<0, la reaction est spontanée, les deux liquides sont donc miscibles
H=0, Cette reaction ne degage/consomme pas d'energie,
S>0, l'entropie du systeme augmente lorsuqe l'on melange les deux solutions.
1.2.1
on pose x(1)+x(2)=1 donc x(1)=1-x(2)
on en deduis
1.2.2
On a un etremale lorsque dG/dx(2)=0
---> x(2)=n(2)/n(1)+n(2)
Je retrouve l'expression de x(2)=n(2)/n(Totale) , ca ne m'avance pas a grand chose
1.2.3 pour x(2) proche de 0, RTn(2)/x(2)>>-RTn(1)/1-x(2), la courbe est decroissante ( de la meme facon que 1/x) et x2 proche de 1 -RTn1/1-x2>>> RTn2/x2, je ne sais pas le dessiner sur ce site mais je pense que j'aurais une courbe similaire a celle de Arc tangente.
2.1
Pour un mélange réelle
2.2
GE=
donc GmE=dG/dn= x(1)RTln(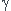 (1)+x(2)RTln((2))
(1)+x(2)RTln((2))
Tu as bien travaillé ! Quelques précisions. Un mélange est considéré comme idéal lorsque les forces d'interactions entre deux molécules 1, entre deux molécules 2 et entre une molécule 1 et une molécule 2 sont identiques, à distances intermoléculaires identiques bien sûr. Les molécules 1 et 2 sont donc interchangeables. Cela explique pourquoi le mélange à T fixe se fait à volume constant et de manière athermique ( V=0 ; H=0). Bien sûr, le désordre à l'échelle moléculaire augmente lors du mélange : l'entropie augmente, l'enthalpie libre diminue. Cela est lié comme tu l'as dis au caractère spontané du phénomène.
1.2.2 :Ton expression le la variation d'enthalpie libre de mélange est correcte mais, pour tracer la courbe, il faut remarquer que n1,n2,x1 et x2 ne sont pas des variables indépendantes. Je ne sais pas trop ce que demande le concepteur de l'énoncé... On peut imaginer la situation suivante : pour une quantité n=n1+n2 fixe (5mol par exemple) on pose n1=n.(1-x2) et n2=n.x2. On obtient alors une courbe correspondant à G=0 pour x2=0 et x2=1 : cas limites où il n'y a pas de mélange. Cette courbe possède un minimum pour x2=x1=0,5 : c'est dans ce cas que le désordre moléculaire est maximum, donc l'entropie de mélange maximale, donc à T fixe....
bonjour, il n'y a pas une facon un peu plus mathematique de montre que le V(m)=0, pour H=0, on etait censé utilise la relation de Gibbs-Helmotz et j'aurais bien aimé savoir comment on aurais fais avec
C'est vrai que j'ai surtout insisté dans mon message précédent sur l'interprétation physique des résultats ...
De même que tu as (voir document fourni pour la démonstration) :
On démontre :
Tu retrouves cela à partir des identités thermodynamiques appliquées avant et après mélange.
Si tu appliques, avant et après mélange la relation de définition de G :
G = H - T.S
tu obtiens, sachant que T reste fixe :
Gmélange=Hmélange - T.Smélange
D'accord, pas besoin d'utilise la relation de Gibbs-Helmotz ( ils le demandent dans l'enonce alors j'aimerais bien comprendre comment faire, j'ai essaye plusieurs choses mais rien n'est bon
Tu y arrives aussi par la relation de Gibbs et Helmholtz , pour un système :
En appliquant cette relation au système après mélange puis au système après mélange et en effectuant une soustraction membre à membre :

